Computational Materials Discovery Goes Platinum
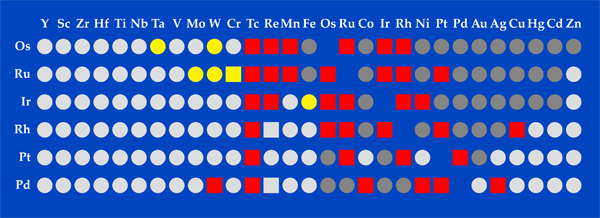
Metal alloys consisting of a platinum group metal—ruthenium, rhodium, palladium, osmium, iridium or platinum—and one or more transition metals are important for catalytic reactions in automobiles, chemistry, and petroleum production. In addition, many of the alloys used in aeronautics and electronics contain platinum group metals. Alloys beyond those currently used in these industries could be of value for many applications, but discoveries have been slow because the costs of trial and error—both in human time and materials—are prohibitive. In Physical Review X, Gus Hart at Brigham Young University, Utah, and colleagues present the largest and most comprehensive computational study of binary metallic platinum group metal alloys [1]. Their computations predict the thermodynamic stability of 28 compounds that haven’t been reported in the literature experimentally. They also predict a few dozen configurations for other alloys, also as yet unreported, that may be experimentally accessible at the nanoscale. The results could thus point to binary phases that might otherwise be overlooked and perhaps lead to a more cost effective use of precious materials.
Matter in new and useful forms will be key for future technologies [2]. But the process to discover, develop, and deploy new materials and related technologies is lengthy, largely because it requires a combination of serendipity, scientific intuition as to which elements to combine, and painstaking experimentation. Typically, a decade or two elapses from the time a new material is discovered to when it can be incorporated into a practical application.
A significant time saving comes from relying more heavily on powerful computers to search for new and improved functional materials. Modern computers can explore the properties of thousands of potentially useful chemical combinations of elements and structures in a fraction of the time that real experiments would take. By organizing the results in a database, researchers can analyze the relationships between multiple materials and discover what are called “property descriptors.” These are quantities that link calculated microscopic parameters (such as formation enthalpies and entropic temperatures) to macroscopic properties (such as stability and formability) and they are the compasses with which researchers navigate the complexity of a multidimensional database [3]. The goal of this approach, called high-throughput materials modeling, is to significantly accelerate the pace of discovery and development of new materials and their assimilation into technology.
High-throughput modeling has a number of benefits. First, scientists can more effectively search for the ideal material for a particular application. Second, since the expected crystal structure and morphology of the materials may be known ahead of time, synthesis is less of a guessing game. Third, researchers can, by comparing large numbers of materials with similar macroscopic properties (such as thermal or electrical conductivity or electromechanical response), discover unexpected descriptors and clarify the origin of emergent functionalities in complex systems. Finally, high-throughput data repositories allow scientists and engineers to easily search for materials as new needs arise: materials found of little promise for applications today may became key to those of tomorrow.
In their new work, Hart et al. considered 153 binary combinations (Fig. 1) of platinum group metals and transition metals. For each combination, they used software called AFLOW, which several of the authors designed for high-throughput computations, and calculated the energies of 250 different possible crystal structures. The calculations allowed the authors to estimate the competition between energy, which favors an ordered compound, and entropy, which favors disordered alloys. AFLOW streamlines the setup of all the input files needed to perform first-principles calculations, controls sources of errors as the calculations are performed on a supercomputer, and facilitates the post-processing and analysis of the data. In total, the authors performed 39,266 calculations, which took 1.82 million CPU hours.
Hart et al. were able to identify crystal structures that experiments have already found and show that other mixtures don’t form stable compounds (again, consistent with experiment). But what is more interesting is that they pinpointed 28 new, so far unexplored, alloys that could potentially lead to new or improved functionalities. The authors don’t explore these functionalities directly, so they remain to be found experimentally.
An important feature of the authors’ work is that they carefully check their calculations against the accessible experimental literature and find agreement in nearly every case. In addition, their predictions are backed by solid thermodynamical analysis. They give detailed information on why certain compounds might have different structures and energetics and why they might have been overlooked in the past. In my opinion, the authors’ approach of providing extensive information across theoretical and experimental research is the best way to lower the communication barrier between subfields and efficiently accelerate the discovery process. This work realizes the decades-old dream of materials scientists to produce computationally enhanced structure maps that integrate experimental and computational data to better guide the search for potentially useful new metal alloys.
Materials scientists may need years to examine all the leads suggested in the study by Hart et al., as some of the materials can only be prepared with highly controlled synthesis, which would make them difficult to deploy into applications. One long-term goal for high-throughput modeling is to design database searches that automatically develop new descriptors for new functionalities. Greater standardization in how materials are cataloged in various databases should be pursued in order to achieve this goal. High-throughput materials modeling has already benefited from new approaches that combine quantum mechanics and thermodynamics, as well as intelligent data mining, so there is every reason to believe these goals are accessible. Already, the materials structure library aflowlib.org [4], which is sponsored by a consortium of universities, national labs, and funding agencies, is an example of an open community that fuels the discovery of new materials.
References
- G. L. W. Hart, S. Curtarolo, T. B. Massalski, and O. Levy, “Comprehensive Search for New Phases and Compounds in Binary Alloy Systems Based on Platinum-Group Metals, Using a Computational First-Principles Approach,” Phys. Rev. X 3, 041035 (2013)
- G. Ceder and K. Persson, “How Supercomputers Will Yield a Golden Age of Materials Science,” Scientific American, Dec 2013
- S. Curtarolo, G. L. W. Hart, M. Buongiorno Nardelli, N. Mingo, S. Sanvito, and O. Levy, “The High-Throughput Highway to Computational Materials Design,” Nature Mater. 12, 191 (2013)
- S. Curtarolo et al. “AFLOWLIB.ORG: A Distributed Materials Properties Repository from High-Throughput Ab Initio Calculations,” Comp. Mater. Sci. 58, 227 (2012)