Breaking down complexity
The expression of genes into proteins is a fundamental and ubiquitous biological process. Since the 1960s [1], we have known that cells also harness this machinery to control complex processes and carry out information-processing tasks, but we are still far from understanding the details of how these behaviors emerge from the intricate interaction of biomolecules within a cell.
One approach to this problem is to assemble a network of biomolecules that functions like the process we want to understand, but is still simple enough that we can write down some equations or rules to describe it. En route to making such molecular networks perform in an external cellular environment (ex vivo), Eyal Karzbrun and colleagues at the Weizmann Institute of Science, Israel, and the University of Minnesota, US, are studying the kinetic details of a basic system, built from parts of E. coli bacteria and reconstituted in a test tube (in vitro), in which genes are expressed as proteins that ultimately degrade. Their work, which appears in Physical Review Letters [2], is part of a growing body of work that advocates building simplified experimental models to better understand biological systems.
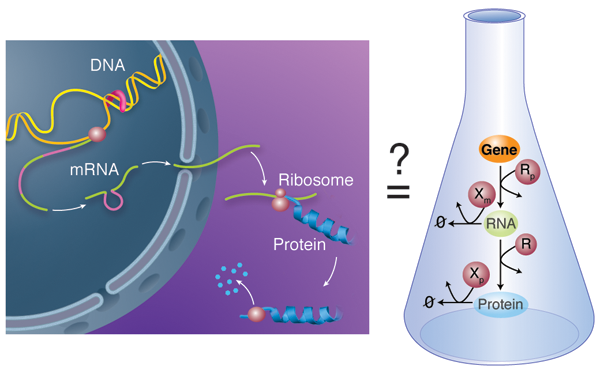
Biological units like cells and multicellular organisms do not act as isolated entities but rather take part in complex networks where each member influences the fate of the others. This is also true below the cellular level: on close examination, the activity of molecular components within a cell is not constant over time, but varies depending on the status of other cellular partners. Chemical processes mediate the interactions within these large, more or less amorphous assemblies called chemical reactions networks. A good example of this concept is the behavior of gene regulatory networks: each individual gene codes for a protein with a relatively simple function; but the network of cross-regulation of all these genes can encode, for example, the incredibly complex developmental process that allows an egg to develop into a full animal [3]. In gene regulatory networks, this regulation can happen at various steps: the transcription of the gene in RNA, the subsequent translation of that RNA into a protein, or the subsequent modifications—and ultimate degradation—of that protein (Fig. 1, left).
Biologists are increasingly focusing on these types of networks. However, even if the intricate structures of more and more of these natural interaction networks are being unraveled, one fundamental issue remains partly unanswered [4]: Is it possible to link a particular network architecture to a specific function? Stated the other way around: Can we reproduce the elaborate dynamics that occur within a cell by assembling an artificial biological network with a consistent network connectivity? Questions like this have given rise to synthetic biology, the learning-by-doing subfield of system biology [5]. The ability to introduce relatively small, exogenous genetic networks in bacteria that then perform a particular function has been fairly successful. The field is still in the early stages of proving itself, however. For example, all attempts to scale up such man-made in vivo constructs beyond five or six interconnections (nodes) between the different parts have been unsuccessful [6]. This is still very far from the hundreds of genes involved in the genetic decision-making processes of even simple organisms.
A possible alternative strategy in figuring out the design rules of reaction networks is to try to build such amorphous systems in a simpler, more controllable context than the way living matter actually does it. In vitro translation systems may provide such an opportunity. These mixtures are obtained either from cell extracts or by reconstituting purified components. They contain all the necessary components for gene-to-protein expression, and as such, provide a platform on which to engineer information-processing genetic networks.
In their work, Karzbrun et al. report the in vitro analysis of the complete chain of events that form the foundation of gene regulatory networks. Specifically, their network includes the process of messenger RNA transcribing the information carried in a single gene and the translation of this information into the corresponding protein (Fig. 1, right). The network also has a mechanism for the messenger RNA and proteins to degrade, meaning they are broken down by enzymes. The team uses classical tools to obtain kinetic information: the incorporation of radio-labeled isotopes to measure RNA kinetics and fluorescent reporting to follow protein translation and degradation. They are also able to decompose the expression of a single gene into its individual steps and study each independently, using purified RNA and protein components. Altogether, they report a quantitative kinetic sketch of all the processes involved, including the degradation pathways.
Although the system they study is a simple form of a network and cannot lead to very complex behaviors, Karzbrun et al.’s modeling approach is notable because it is based on kinetic parameters that they are able to experimentally—and relatively easily—measure. In contrast, because it is often difficult to obtain accurate kinetic and thermodynamic values describing biological processes within a cell, the mathematical analysis of in vivo molecular networks generally rely on rather crude estimations of the various parameters, or even ad hoc assumptions about the mathematical expressions that best describe the chemical processes [3]. One example of the sort of information Karzbrun et al. are able to obtain from their system relates to the effects of protein degradation. The team genetically tagged their fluorescent protein with a small sequence to force it to follow a specific degradation pathway. (During the expression process, this sequence results in an extra peptide called the ssrA tag, which designates the protein as the target of an enzyme that destroys proteins). This is a rather classic tool of synthetic biology for increasing the dynamic of artificial in vivo networks by decreasing protein lifetime. However, the authors report here that this process is extremely efficient: the machinery involved saturates for a low level of tagged proteins and the degradation rate is independent of the concentration of this target (what is called zeroth-order kinetic). It is important to realize that the way a protein dies is as important as the way it is synthesized for defining the global behavior of a dynamic system. In this specific case, the zeroth-order degradation (which had already been noted in an in vivo study [7]), basically controls the global system: Whereas one would intuitively expect such a production/degradation system to robustly reach a steady state (which is the most interesting behavior that can be expected in this case), Karzbrun et al. show instead that this steady-state solution doesn’t exist for a large area of the system’s parameter space, a result that may have strong consequences in future attempts to build networks with more complex dynamics.
One can see another advantage to the use of in vitro systems for the engineering of molecular networks: they are free of stochasticity. Indeed, many biological processes are stochastic. (This is particularly the case in gene expression in a cell, since this process uses molecular components that are present in low numbers in a cell.) While this is a fundamental characteristic of life, with far reaching consequences [8], it can also become a burden when it comes to building a specific robust dynamic behavior. Bulk in vitro systems may help distinguish between the behaviors that are direct consequences of cell stochasticity and those that are not. In the future, one may think of compartmentalization of in vitro systems as a means to reintegrate stochasticity, in a controlled manner.
The next step for this and related work will be to demonstrate that complex behaviors, similar to what is observed in vivo, can be obtained using in vitro expression systems as the machinery to perform genetically encoded networks. Recently, other bio-inspired, but also in vitro, approaches have permitted the rational design of artificial switches or even oscillators [9] that mimic their biological counterparts. These various approaches will probably change our vision of out-of-equilibrium chemical systems and the information they contain. They will offer a better understanding of how biological behaviors, and ultimately life, may emerge from complex reaction networks.
References
- F. Jacob and J. Monod, J. Mol. Biol. 3, 318 (1961)
- E. Karzbrun, J. Shin, R. H. Bar-Ziv, and V. Noireaux, Phys. Rev. Lett. 106, 048104 (2011)
- E. H. Davidson, Nature 468, 911 (2010)
- W. Ma, A. Trusina, H. El-Samad, W. A. Lim, and C. Tang, Cell 138, 760 (2009)
- T. S. Gardner, C. R. Cantor, and J. J. Collins, Nature 403, 339 (2000)
- P. Purnick and R. Weiss, Nat. Rev. Mol. Cell Biol. 10, 410 (2009)
- W. W. Wong, T. Y. Tsa, and J. C. Liao, Mol. Syst. Biol. 3, 130 (2007)
- T. J. Perkins and P. S. Swain, Mol. Syst. Biol. 5, 326 (2009)
- J. Kim and E. Winfree, Mol. Syst. Biol. (to be published); K. Montagne, R. Plasson, Y. Sakai, T. Fujii, and Yannick Rondelez, (to be published)