Coherent Control of Chemical Reactions on the Attosecond Time Scale
Since the invention of lasers, people have dreamed of employing the laser as a “photonic reagent” to steer the outcome of a chemical reaction, using light to selectively cleave or form a chemical bond in a polyatomic molecule, or to vary the branching ratios of a photoinduced chemical process. This fascinating possibility has both fundamental and practical implications: on the one hand, it allows a better understanding of the underlying atomic and molecular physics; on the other hand, it may provide a way to synthesize chemical substances with higher efficiencies, while at the same time reducing unwanted by-products, or even promote entirely new reaction pathways allowing synthesis of compounds for which no conventional methods exist [1]. Reporting in Physical Review Letters, Xinhua Xie at the Vienna University of technology, Austria, and co-workers advance this research by extending approaches recently developed for atomic systems and simple molecules into the realm of polyatomic molecules [2].
The path to this result has been a difficult one. The first implementations of laser selective photochemistry used narrow-bandwidth light tuned in resonance with the vibrational transition of a specific chemical bond as molecular-scale “scissors,” depositing energy into that mode until the bond breaks. This approach failed, however, due to the occurrence of intramolecular vibrational energy redistribution, which rapidly transfers energy from the targeted mode to others in the molecule, thus spoiling the modal selectivity.
Subsequent approaches exploited the quantum nature of light-matter interaction in order to design a suitably tailored light field capable of steering the molecule towards the desired photoproduct. The so-called Brumer-Schapiro scheme [3] uses two monochromatic laser fields with commensurate frequencies and exploits quantum-mechanical constructive and destructive interferences between different excitation pathways leading to the same final state. By properly controlling amplitudes and phases of the fields, it is possible to selectively enhance one reaction channel while suppressing the others. The “pump-dump” scheme, proposed by Tannor, Kosloff, and Rice [4], is based on two time-delayed excitation pulses. The first “pump” pulse creates an electronic wave packet on the excited-state electronic potential energy surface, which then evolves freely until the “dump” pulse brings it down to the ground state along the desired reaction channel, allowing the wave packet to “jump” any barrier obstructing that channel.
Both approaches, however, use only a limited number of degrees of freedom, such as the phase difference between the laser fields and the time delay between pump and dump pulses. In a generalization of the method, Rabitz and co-workers [5] proposed the use of an ad hoc laser pulse, specifically designed for the molecule, in order to enhance a given reaction channel while suppressing the others. Such an optimum pulse generally has a complex temporal profile, with tailored amplitudes and phases of its different spectral components, which can be straightforwardly generated using modern pulse-shaping technologies. It was found, however, that this approach to coherent control generally fails, because the potential energy surface of the molecule is not known with sufficient accuracy to allow reliable design of the optimal control pulse. This problem was solved by a “closed-loop” method, in which one lets the molecule “solve its own Schrödinger equation” by using the output of the experiment to feed a learning algorithm that generates the optimum pulse profile in an iterative optimization cycle [5]. Using this approach, breakthrough results were achieved in coherent control of chemical reactions [6] and in the control of the efficiency of photoinduced processes in photosynthesis [7] and vision [8].
More recently, researchers have focused on a new degree of freedom called the carrier-envelope phase (CEP), which provides an additional knob for coherent control. CEP is defined as the phase delay between the tallest half-cycle of the electric field under the pulse envelope and the peak of the envelope itself; its stabilization and control allow the synthesis of not only the intensity profile but also the electric field of the light wave form. Obviously, the CEP becomes particularly relevant when the pulse envelope encompasses only a few cycles of the carrier wave, as is the case for infrared pulses with duration of the order of a few femtoseconds. The CEP has been demonstrated to play a crucial role in controlling strong-field laser-matter interactions, allowing the generation and measurement of reproducible isolated attosecond pulses [9] and the manipulation of electron emission from atoms [10]. The subcycle evolution of the electric field of few-cycle laser pulses has also been exploited to achieve controlled motion of bound electrons during dissociation of simple diatomic molecules [11].
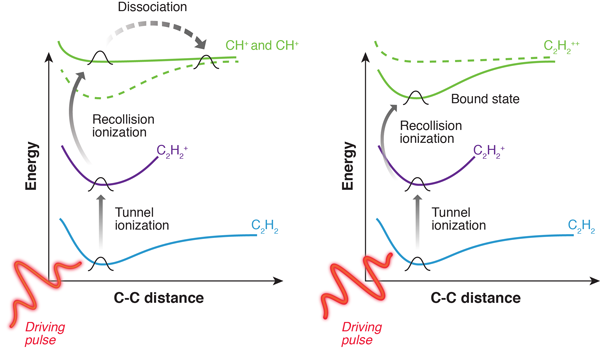
Xie and co-workers extend this approach to more complex polyatomic molecules (Fig. 1), demonstrating experimentally how the photofragmentation process can be selectively triggered by CEP-controlled few-cycle pulses [2]. This result provides a completely new and powerful approach to coherent control, which relies on subcycle electron wave-packet dynamics.
To study the photofragmentation process in the molecular frame, the authors employed a method based on ion-electron coincidences, in which the orientation of the molecule at the time of ionization is reconstructed from the momenta of electron and ion. This approach, known as cold target recoil ion momentum spectroscopy, combines electrostatic and magnetic fields to guide electrons and ions onto position-sensitive detectors on either side of the interaction region [12]. Suitable algorithms then allow the original momentum of all charged particles to be reconstructed from the time of arrival and the position on the two detectors. The experiments have been performed on different polyatomic molecules, namely acetylene, ethylene, and butadiene, strongly driven by few-optical-cycle infrared pulses. For all these species, the fragmentation yields were very strongly modulated by varying the CEP.
By combining the experimental observations with a simple theoretical model the authors were able to identify the mechanism responsible for the strong control of fragmentation yields on the subcycle time scale (Fig. 1). The CEP dependence is found to be largely independent of the molecular sample, and is related to the process that populates the dissociative states involved in the photofragmentation. For these reasons, the underlying mechanism has been recognized as recollision ionization: photoexcitation first ionizes the molecule through tunnel ionization (the laser field modifies the binding potential, allowing an electron to tunnel out). Following tunnel ionization, the photogenerated electron is driven by the light field to recollide with the parent ion. Since the motion of the photogenerated electron wave packet is directed by the external driving field, the exact phase of the field can determine what happens after recollision, thus encoding the information about the CEP. For CEP values resulting in sufficiently high kinetic energy of the recolliding electron, recollision ionization is able to remove an electron from an inner shell, bringing the molecule into a dissociative excited state and promoting photofragmentation. On the other hand, for those CEP values resulting in lower recollision energy, ionization only removes an electron from the highest occupied molecular orbital, thus bringing the molecule to a nondissociative state and preventing photofragmentation (see Fig. 1).
This result is both surprising and fascinating since it paves the way to selective photochemistry on complex molecular systems, with possible important applications in light-assisted synthesis of molecules by controlled photofragmentation reactions. At the same time, the paper provides a very elegant fundamental study of strong-field light-matter interaction, showing the new physics that can be accessed when ultrashort light pulses manipulate electron trajectories on their elementary time scales.
References
- H. Rabitz, R. de Vivie-Riedle, M. Motzkus, and K. Kompa, “Whither the Future of Controlling Quantum Phenomena?” Science 288, 824 (2000)
- X. Xie et al., “Attosecond-Recollision-Controlled Selective Fragmentation of Polyatomic Molecules,” Phys. Rev. Lett. 109, 243001 (2012)
- P. Brumer and M. Shapiro, “Laser Control of Molecular Processes,” Annu. Rev. Phys. Chem. 43, 257 (1992)
- D. J. Tannor, R. Kosloff, and S. A. Rice, “Coherent Pulse Sequence Induced Control of Selectivity of Reactions: Exact Quantum Mechanical Calculations,” J. Chem. Phys. 85, 5805 (1986)
- R. S. Judson and H. Rabitz, “Teaching Lasers to Control Molecules,” Phys. Rev. Lett. 68, 1500 (1992)
- A. Assion, T. Baumert, M. Bergt, T. Brixner, B. Kiefer, V. Seyfried, M. Strehle, and G. Gerber, “Control of Chemical Reactions by Feedback-Optimized Phase-Shaped Femtosecond Laser Pulses,” Science 282, 919 (1998)
- J. L. Herek W. Wohlleben, R. J. Cogdell, D. Zeidler, and M. Motzkus, “Quantum Control of Energy Flow in Light Harvesting,” Nature 417, 533 (2002)
- V. I. Prokhorenko A. M. Nagy, S. A. Waschuk, L. S. Brown, R. R. Birge, and R. J. D. Miller, “Coherent Control of Retinal Isomerization in Bacteriorhodopsin,” Science 313, 1257 (2006)
- A. Baltuška et al., “Attosecond Control of Electronic Processes by Intense Light Fields,” Nature 421, 611 (2003)
- G. G. Paulus, F. Lindner, H. Walther, A. Baltuška, E. Goulielmakis, M. Lezius, and F. Krausz, “Measurement of the Phase of Few-Cycle Laser Pulses,” Phys. Rev. Lett. 91, 253004 (2003)
- M. F. Kling et al., “Control of Electron Localization in Molecular Dissociation,” Science 312, 246 (2006)
- J. Ullrich, R. Moshammer, A. Dorn, R. Dörner, L. Ph H. Schmidt, and H. Schmidt-Böcking, “Recoil-Ion and Electron Momentum Spectroscopy: Reaction-Microscopes,” Rep. Prog. Phys. 66, 1463 (2003)