Proteins Hook up Where Water Allows
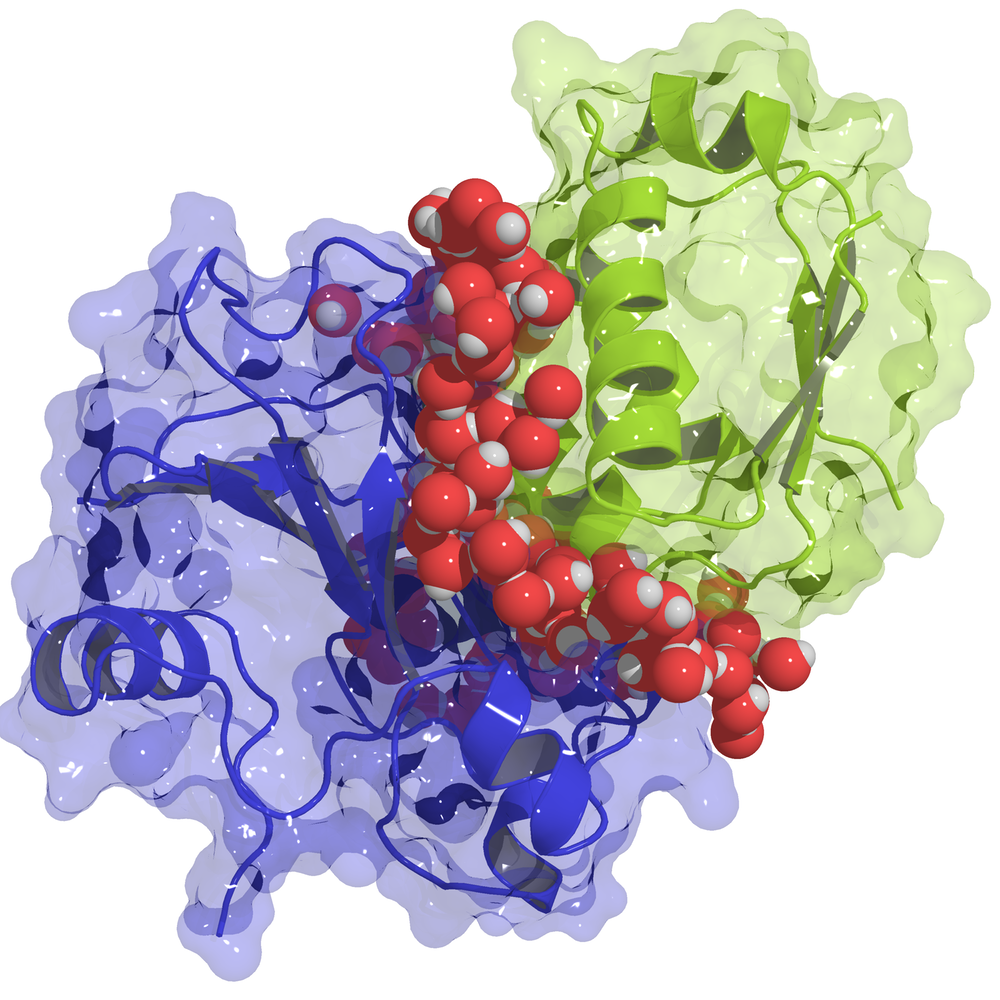
Many biological processes depend on the physical attraction between pairs of proteins. For proteins that float freely in a cell, this attraction depends strongly on the arrangement of a surrounding “shell” of water molecules. In Physical Review Letters, a researcher shows that his calculations can predict the locations of strong binding on surfaces of proteins by finding the regions where the ordinary network of water molecules is disrupted. If the calculations can be streamlined, they may help researchers develop small-molecule drugs that work by modifying the binding to block biological activity.
Proteins do most of the work in biology, often by working in pairs or groups. For example, growth hormone in the bloodstream communicates a “grow” message to tissues by binding to specific receptor proteins in the tissues. Other proteins assemble into multi-molecule machines called complexes, such as the proteasome that digests cellular trash. To perform these tasks, proteins must “recognize” potential binding partners based on their complementary shapes and their chemical properties. But “people don’t know how to predict interactions” of proteins from their structures, says Ariel Fernández of the Argentine National Research Council in Buenos Aires, the University of Chicago, and the Morgridge Institute in Madison, Wisconsin.
In previous experiments, drug researchers compared the strength (energy) of binding of a protein pair, such as human growth hormone and its receptor, with that of engineered versions of the pair having specific mutations. A protein is a long, folded chain of various amino acids. By painstakingly replacing one amino acid at a time, and checking the binding energy for each mutant version of the protein, the researchers flagged “hot spots” in the structure where a mutation had the strongest influence on binding. So these locations appeared to be essential for the strong protein-protein bond.
Fernández explains these hot spots as regions where the surrounding water molecules can’t form the network of hydrogen bonds that exists in ordinary water, so they are easily displaced in the protein binding process. Researchers have long ascribed some of the strength of binding to the fact that some amino acids are hydrophobic (“water avoiding”) and thus prefer contact with another protein over exposure to water. But this description doesn’t account for important features, such as the effect of neighboring amino acids and the ability of the water molecules to penetrate into the protein structure, says Fernández. “You have to extend the notion of hydrophobicity to the nanoscale,” he says, which is “a very different creature.”
To quantify the water structure near a protein surface, Fernández came up with a new way to calculate the average number of neighbors for each water molecule at each position. In normal water, each water molecule makes a hydrogen bond with four neighbors—it has a “coordination” of four. Near a protein, the coordination drops, but only gradually. Fernández started with the detailed structures for nine proteins that form pairs and calculated the expected coordination at each location around the proteins. He found that the regions where the coordination is significantly less than four matched well with the hot spots. This result supported the idea that in these regions it takes less energy to dislodge water molecules to allow another protein to approach. Overall, the coordination explained much of the measured binding energy, but not all of it.
The current version of the calculation is computationally expensive, so further refinements will be needed before it can be widely used to predict binding. But Fernández hopes it can help researchers devise drugs that interfere with the process, noting that “disrupting protein-protein interfaces is the holy grail” for pharmaceuticals.
Pablo G. Debenedetti of Princeton University agrees that this “solvent-centric” approach appears to predict the experimental hotspots. “In biology you have to be careful with simple overriding principles,” he says, “but this is very promising.”
–Don Monroe
Don Monroe is a freelance science writer in Murray Hill, New Jersey.