Controlling a Molecule’s Fate
Ethylene is a small hydrocarbon molecule ( ) that plays numerous roles in our daily lives. As a plant hormone, it regulates growth and can be used to promote the ripening of fruits. As a highly reactive molecule, it serves as a major building block in the chemical industry, where the molecule is often polymerized to make plastics or oxidized for the manufacture of detergents and automotive antifreeze. The underlying physics that drives ethylene’s functionality is the ultrafast motions of the electrons and nuclei that constitute the molecule. A better understanding of how these motions depend on each other could allow greater control of the behavior of ethylene or any other molecule. Writing in Physical Review X, Xinhua Xie, from Vienna University of Technology in Austria, and colleagues have performed experiments that reveal the correlated electron-nuclear dynamics and the possibility of steering the breaking of the ethylene molecule by an ultrashort laser pulse [1].
Recent work has demonstrated how ultrashort laser pulses can control the chemical dynamics of the small hydrocarbon molecule, acetylene ( ) [2]. In this case, the phase and amplitude of the laser pulses were tuned through adaptive feedback, based on the three-dimensional momentum of the resulting molecular fragments. This self-learning closed-loop routine turns the laser pulse into a molecular-photo-scissor, which can snip the molecule apart in different ways [3]. However, it is difficult to interpret the algorithm’s complicated output of phase and envelope information and relate it to physical interactions between the laser and the molecule. The complexity dramatically increases when these laser methods are employed for larger, polyatomic molecules.
A more direct and efficient method for the control of the molecular dynamics is one based on the physical understanding of the correlated electron-nuclear dynamics that occur on the time scales of ultrashort laser pulses. Given their difference in mass, the electrons and nuclei in a molecule move on time scales of attoseconds ( ) and femtoseconds ( ), respectively. This factor of difference means that a pulse from a laser can excite the electrons (e.g., freeing them into the continuum) before the nuclei have had time to fully respond. In the case of photoionization, the extremely fast escape of the electron alters the potential energy of the molecule almost instantaneously, creating a new potential energy surface on which the nuclei subsequently move. The final state—whether it is a reformed molecule, a bond breakage, or a chemical reaction—depends on which electron is excited. The fate of the molecule is therefore electronically predetermined.
Molecules have many electrons that can interact with an intense laser field, but typically only certain electrons are likely to be photoionized. The most susceptible electrons are those in the highest occupied molecular orbital (HOMO). Beyond that, there is a significant probability for electrons to be freed to the continuum by a strong laser field from more tightly bound, lower-lying orbitals (HOMO-1 or HOMO-2) of the molecule [4,5]. Some of these electrons are responsible for binding nuclei together, so if they are knocked out by laser light, the molecular bond is weakened or broken. It is not so easy to target specific electrons with a laser pulse, but certain parameters allow some selectivity. For example, the geometric shape of different orbitals means that one can increase or decrease the probability of an electron tunneling out of a particular orbital by changing the laser intensity and/or orientating the molecule relative to the laser polarization [6]. In addition, the duration of the pulse provides another controllable parameter in that a longer pulse can give more time for “two-step” processes that rely on correlated motions of electrons and nuclei at various time scales.
Xie and co-workers investigated whether the breakup of ethylene could be controlled by varying the intensity and duration of an ionizing laser pulse [1]. In their experiment, laser pulses interacted with a jet of ethylene molecules in an ultrahigh-vacuum apparatus. The team looked specifically at interactions in which two electrons are removed, followed by the doubly ionized molecule breaking up into two ion fragments. These double ionizations allow both fragments to be observed with cold target recoil ion momentum spectroscopy [7]. In this setup, a spectrometer with a time- and position-sensitive microchannel plate detector identified coincident ion pairs. Using the measured times-of-flight and positions of the ions on the plate detector, the team reconstructed the three-dimensional momenta that the fragments had at the moment when they split from the molecule. The researchers then calculated the ejection directions and kinetic energies of the fragments to reveal the nuclear dynamics determined by the electron excitation.
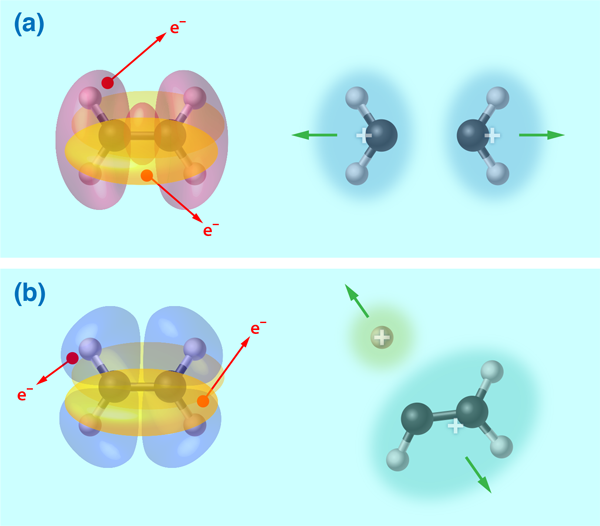
By comparing the experimental observations with the geometric profiles of the orbitals and the calculated potential energy surfaces of the doubly ionized ethylene molecule, the authors connected the dots between the initial electron ionizations and the final ion states. For example, they established that the symmetric breaking of the ethylene into two ions is dominated by removing an electron from the tightly bound HOMO-2 orbital along the C–C bond [Fig. 1(a)]. In contrast, the deprotonation and isomerization-accompanied breaking of the ethylene into ( ) and ( ) fragment pairs were dominated by removing one HOMO electron and one HOMO-1 electron [Fig. 1(b)]. With increasing laser intensity, additional pathways for the symmetric breakup channel of the doubly ionized ethylene were opened up by either directly removing two HOMO-2 electrons or through a two-step process in which the laser pulse first removes two electrons and then subsequently excites the doubly ionized molecule into an excited state that later splits into two ion pairs. As compared to the other two breakup channels, the symmetric breaking of the ethylene by ionizing the tightly bound HOMO-2 orbital is evidently favored at high laser intensity. Moreover, the ethylene is found to be more susceptible to breakage when the laser pulses are made shorter in duration.
These results demonstrate that the breaking of ethylene is electronically predetermined and is controllable by varying the intensity and temporal duration of an ultrashort laser pulse. The progress displayed here is significant since it not only brings us a new understanding of the correlated electron-nuclear dynamics of a widely used polyatomic hydrocarbon molecule, but it also provides us an efficient approach for controlling chemical dynamics by selectively exciting electrons from various orbitals at the very beginning. Going forward, researchers may improve the performance of this routine by spatially aligning molecules with a preliminary pulse of weak (nonionizing) light [8]. Such prealignment allows better control of which electron orbitals will be targeted by the subsequent ionizing pulse.
References
- Xinhua Xie et al., “Electronic Predetermination of Ethylene Fragmentation Dynamics,” Phys. Rev. X 4, 021005 (2014)
- E. Wells et al., “Adaptive Strong-Field Control of Chemical Dynamics Guided by Three-Dimensional Momentum Imaging,” Nature Commun. 4, 2895 (2013)
- A. Assion, T. Baumert, M. Bergt, T. Brixner, B. Kiefer, V. Seyfried, M. Strehle, and G. Gerber, “Control of Chemical Reactions by Feedback-Optimized Phase-Shaped Femtosecond Laser Pulses,” Science 282, 919 (1998)
- H. Akagi, T. Otobe, A. Staudte, A. Shiner, F. Turner, R. Dörner, D. M. Villeneuve, and P. B. Corkum, “Laser Tunnel Ionization from Multiple Orbitals in HCl,” Science 325, 1364 (2009)
- J. Wu, L. Ph. H. Schmidt, M. Kunitski, M. Meckel, S. Voss, H. Sann, H. Kim, T. Jahnke, A. Czasch, and R. Dörner, “Multiorbital Tunneling Ionization of the CO Molecule,” Phys. Rev. Lett. 108, 183001 (2012)
- X. M. Tong, Z. X. Zhao, and C. D. Lin, “Theory of Molecular Tunneling Ionization,” Phys. Rev. A 66, 033402 (2002)
- R. Dömer, V. Mergel, O. Jagutzki, L. Spielbergeri, J. Ullrich, R. Moshammer, and H. Schmidt-Böcking, “Cold Target Recoil Ion Momentum Spectroscopy: A ‘Momentum Microscope’ to View Atomic Collision Dynamics,” Phys. Rep. 330, 95 (2000)
- H. Stapelfeldt and T. Seideman, “Aligning Molecules with Strong Laser Pulses,” Rev. Mod. Phys. 75, 543 (2003)