Waiting for the Quantum Simulation Revolution

Quantum computers are hot. The US Congress has authorized up to $1.2 billion of research funding for quantum information science, including computing. Some of the world’s largest technology companies are rapidly building prototype devices, with Google reportedly documenting the first case of a quantum computer that can beat classical computers at a specific task just last month.
One of the most anticipated uses for these machines is as a tool for developing new drugs, catalysts, and materials. As a 2017 headline in Chemical and Engineering News put it, “Chemistry Is Quantum Computing’s Killer App.” Statements from business and political leaders have also helped to fuel the excitement. An IBM vice president predicted quantum-computer-enabled materials discovery in three to five years. And Lamar Smith, the recently retired Congressman who championed the quantum funding bill, promised “drastic improvements in … the development of new medicines and materials.” He also touted a quantum computing industry “just over the horizon.”
Such hopes ride on quantum computers’ potential to exactly simulate chemicals’ and materials’ quantum structures and behaviors. But, for all their progress, the devices still need major advances to become practical. Materials scientists and chemists caution that quantum machines are far from competing with today’s increasingly powerful classical simulation methods. The technology’s ability to make a near-term impact “is way overhyped currently,” says Kristin Persson, a materials scientist at Lawrence Berkeley National Laboratory in California.
Quantum computing experts acknowledge that the technology has a long way to go but say they are making rapid progress in both hardware and software development. They also point to specific applications at which classical simulations struggle and quantum machines could have their first impacts. And, they argue, clever design choices may ultimately allow the machines to work in tandem with classical computers, maximizing the benefits of each.
Theoretical beginnings, practical advances
Quantum computers use quantum bits, or qubits, which can store the values zero, one, or a combination of both—for example, 75% one and 25% zero. Moreover, qubits can be quantum mechanically entangled with one another. These properties enable quantum computers to rapidly explore a wide range of possible solutions to certain classes of problems.
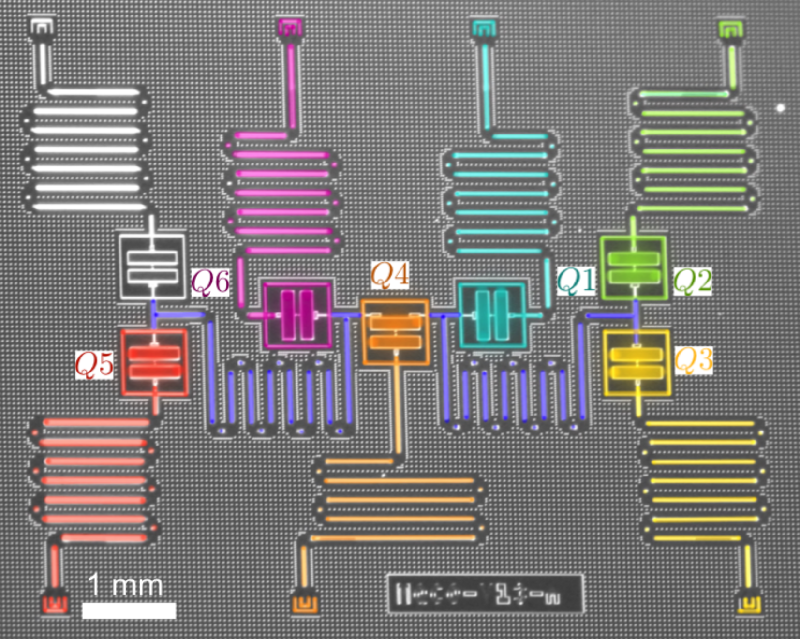
The field of quantum computing was launched in 1981, when Richard Feynman, speaking at the Massachusetts Institute of Technology, pointed out that a quantum computer could, in theory, simulate a molecule or material exactly, rather than just approximately. At the time, it was purely a thought experiment. But by the mid-1990s, physicists had produced qubits and demonstrated quantum logic gates, which are the building blocks of computer operations. The quantum states of these qubits were easily disturbed by interactions with the environment—a formidable challenge that quantum-computer makers have struggled with ever since. Today, rudimentary quantum computers consisting of up to dozens of interconnected qubits can crunch through 100 or so gate operations before crashing. Caltech physicist John Preskill has termed this type of operation “noisy intermediate-scale quantum computing” [1].
Feynman’s proposals are no longer just theoretical. In 2016, Google announced that it had simulated the hydrogen molecule and estimated its ground-state energy using a quantum computer built around tiny superconducting circuits [2]. IBM did the same for lithium hydride and beryllium hydride in 2017 [3]. And earlier this year, ionQ, a startup company in College Park, Maryland, simulated water, the largest molecule to date, using a computer in which trapped ions served as the qubits [4].
Such demonstrations are “a huge step,” says Markus Reiher, a theoretical chemist at the Swiss Federal Institute of Technology (ETH) in Zurich. “Ten years ago, nobody would have guessed that we would see these quantum computing results.”
But Reiher also notes that the recent simulations fell well short of what classical computers can do. The quantum computers couldn’t represent all of the electron orbitals in each molecule, which would be necessary for an exact ground-state energy calculation. Even for the hydrogen molecule, an exact energy calculation would require 56 qubits, Reiher says; the ionQ computer used only four.
Chris Monroe, a physicist at the University of Maryland, College Park, and a founder of ionQ, agrees that his team’s simulation of water did not match classical machines’ accuracy. But he says that demonstrations that do so are “within reach” and simply require marshalling more qubits and running algorithms for longer. And he notes that one does not have to add many atoms to get to molecules whose ground-state energies classical methods cannot calculate precisely. Even a compound as simple as caffeine, with just 24 atoms, would be new territory. “When you have 50 to 100 electrons [being simulated by a quantum computer], people will start to take notice,” Monroe predicts.
80-year head start for classical computers
For now, classical computer simulation remains king. That’s thanks to an almost eight-decade history, going back to World-War-II-era Monte Carlo simulations of nuclear detonations. The 1950s saw the development of molecular dynamics techniques, which treat atoms as balls and chemical bonds as springs. And in the 1960s, physicists developed density functional theory (DFT), a method to approximate the quantum interactions of electrons within molecules or materials.

Today chemists and materials scientists can run molecular dynamics simulations of millions of atoms, though without an explicit representation of individual electrons. And thanks to large supercomputers, researchers using DFT can approximate the electronic structures of complicated molecules and materials with thousands of atoms.
Persson, for example, has used DFT to simulate possible cathode and electrolyte materials for a new kind of battery that would use magnesium ions, rather than lithium ions, as charge carriers. Researchers produced her most promising candidates in the lab and confirmed that they have the properties she predicted.
Persson, like everyone interviewed for this story, would enthusiastically embrace quantum computers that could supercharge her simulations. But as a materials expert, she is keenly aware of how long it can take to discover novel materials such as superconductors that would enable Google, IBM, and others to maintain quantum states for longer times. Persson expects to continue relying on classical methods for the foreseeable future. “[For] the kinds of computations I do, [quantum computers] are not even close,” she says. “We’re talking 10 years at least.”
Emily Carter, a theoretical chemist at the University of California, Los Angeles, says quantum computing may help in studying materials whose complex electronic structures strain existing classical methods, such as superconductors in which individual electron-electron interactions determine the material’s behavior. If researchers can develop quantum algorithms for simulating such “strongly correlated” materials, “you could end up learning things that are qualitatively new,” she says.
{kind=link}
Existing classical methods also struggle at estimating the rates of chemical reactions, which are highly sensitive to even small errors in molecular energy calculations, says Ryan Babbush, a quantum algorithm specialist at Google. He envisions DFT continuing to dominate molecular-structure calculations, but quantum computers could start to handle reaction mechanisms and rates, where accuracy is paramount, he says. “One really should not think of methods like DFT as being in competition with quantum computing, because they are trying to target a fundamentally different level of accuracy and usually for a different purpose.”
Computers on drugs
Among the areas where quantum computers may have received the most hype is the multibillion-dollar industry of drug discovery. A 2018 report by the Boston Consulting Group suggested that a massive $20-billion quantum pharmaceutical industry could emerge by 2030.
Pharmaceuticals are typically small molecules of 50 to 80 atoms. But to be effective, drugs must interact with biological molecules such as proteins, which can contain thousands of atoms, far beyond what any quantum computer will be able to handle in the near future. “If you want to bring [quantum computers] to the point where they can really compete with the methods we have nowadays, the gap is still significant,” says Marco de Vivo, a theoretical biochemist at the Italian Institute of Technology in Genova, referring to current methods such as molecular dynamics and DFT.
True, says Google’s Babbush. But the noisy quantum processors of the near future need not tackle an entire protein to have an impact. “We could use a classical method such as DFT to treat the vast majority of the system and then treat just the most quantum part—for example, the electrons involved in forming or breaking a bond between the protein and ligand—on the quantum computer,” he says. De Vivo also sees promise for quantum computers in another role: rapidly screening large numbers of molecules to more efficiently pick out promising drug candidates for further study.
Quantum computers for simulating catalysis
Many scientists believe that the first impact of quantum computers could be in catalysis research—the search for compounds that facilitate important chemical reactions. In 2017, Reiher and a team of Microsoft researchers studied a concrete use case: They investigated how large a quantum computer would need to be to resolve the energy states of FeMoco, a crucial part of the catalyst that allows bacteria to convert molecular nitrogen from the air into forms accessible to plants. Chemists hope that understanding FeMoco could accelerate efforts to improve upon the artificial catalyst currently used in industrial fertilizer production—a process that accounts for several percent of the world’s energy consumption. Unfortunately, classical methods such as DFT struggle to resolve the many closely-spaced energy levels of the complex FeMoco molecule.
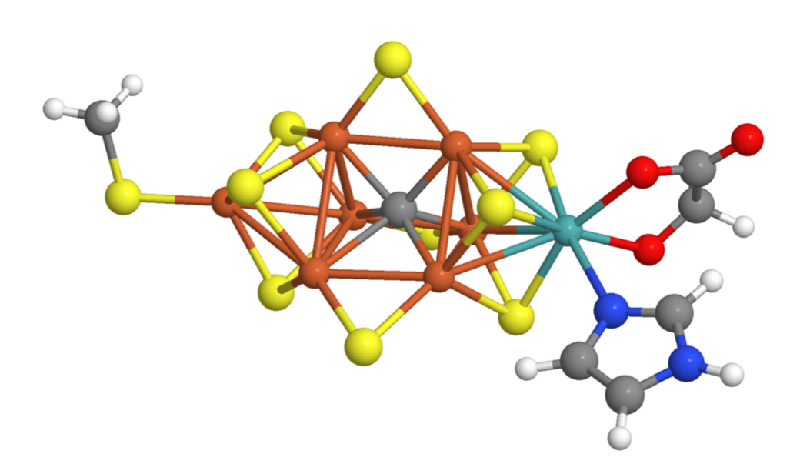
Reiher and his colleagues calculated that a quantum computer with around 200 perfectly-operating qubits could solve the FeMoco problem in a matter of weeks or months [5]. Such a short time, says Reiher, was a surprise. However, qubits don’t operate perfectly, so thousands of additional qubits would likely be needed to stabilize the quantum states of the 200 “logical” qubits, he says. The largest publicly-known, logic-gate-based quantum computer, produced by Google, has 72 qubits, and they are far from perfect; their fragile quantum states collapse in less than a millisecond. Moreover, Reiher adds, classical simulation techniques are continuing to improve, so they are “a moving target.”
Software and hybrid machines
Quantum computer advocates acknowledge that they have work to do. But they say they have an ace up their sleeve that could help quantum computing progress more rapidly than skeptics may think: quantum software development. Microsoft researchers, for example, used algorithm improvements to reduce by a factor of ten million the number of quantum logic operations needed to exactly solve FeMoco. “On the software side, we’re not waiting for this machine to be perfect,” says Alán Aspuru-Guzik, a theoretical physicist at the University of Toronto who developed the molecular simulation algorithm used by Google, IBM, and ionQ.
Ultimately, researchers say, the quantum computer will probably never be a one-stop-shop for materials or chemical discovery. Instead, it may be part of a workflow in which a classical computer sets up the problem and feeds it to the quantum machine for specific computational steps—or vice versa. “Maybe I use the quantum computer to calculate 100 molecules, fit the machine learning model, fix DFT and then use a classical computer from there,” says Aspuru-Guzik.
When will quantum simulations transform chemistry and materials science?
After the push to secure Congressional research funding last year, some quantum computing experts have been trying to bring the promises back to Earth. In an essay published this spring in VentureBeat, Monroe cautioned that “too much hype risks disillusionment that may slow the progress.” He predicted that 5 to 10 years of additional research and development will be needed before quantum computers start solving useful problems.
Several materials scientists and chemists interviewed for this story viewed 10 years as the lower bound for when a quantum computer could become useful. “In my community, people are skeptical,” says Reiher. “If you’re conservative, give it another 20 years.” But, he adds, a mature quantum computer would revolutionize theoretical chemistry. “If I see that that hardware is coming, I will change the whole research program of my group,” he says.
– Gabriel Popkin
Gabriel Popkin is a freelance science writer in Mount Rainier, Maryland.
References
- J. Preskill, “Quantum computing in the NISQ era and beyond,” Quantum 2, 79 (2018).
- P. J. J. O’Malley et al., “Scalable quantum simulation of molecular energies,” Phys. Rev. X 6, 031007 (2016).
- A. Kandala et al., “Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets,” Nature 549, 242 (2017).
- Y. Nam et al., “Ground-state energy estimation of the water molecule on a trapped ion quantum computer,” arxiv.org/1902.10171.
- M. Reiher, N. Wiebe, K. M. Svore, D. Wecker, and M. Troyer, “Elucidating reaction mechanisms on quantum computers,” Proc. Natl. Acad. Sci. USA 114, 7555 (2017).