Chemistry Nobel Awarded for an AI System That Predicts Protein Structures
The Nobel prize for Chemistry has been awarded this year for work on the computational design and structural prediction of protein molecules. Half of the prize goes to David Baker of the University of Washington and half jointly to Demis Hassabis and John M. Jumper of the artificial intelligence (AI) company Google DeepMind, based in London.
With the 2024 physics Nobel Prize given to researchers for their foundational work on the neural networks used by today’s AI systems, the chemistry prize further emphasizes the transformative effect such machine learning algorithms are having in many fields. The chemistry award “recognizes how AI is supercharging science,” says Frances Arnold of the California Institute of Technology, a chemistry Nobel laureate for her work on creating non-natural enzymes with new functions.
Hassabis and Jumper were the key architects of an algorithm called AlphaFold that can predict the structure of a protein purely from knowledge of the sequence of amino acids linked together in its molecular chain. Baker shares the award for his work on designing and synthesizing new, non-natural protein structures from scratch. That task is now greatly assisted by AlphaFold, which can, in effect, be run in reverse to predict the sequence that will fold into a given target structure.
All proteins are composed of chains of amino acids, which generally fold up into compact globules with specific shapes. The folding process is governed by interactions between the different amino acids—for example, some of them carry electrical charges—so the sequence determines the structure. Because the structure in turn defines a protein’s function, deducing a protein’s structure is vital for understanding many processes in molecular biology, as well as for identifying drug molecules that might bind to and alter a protein’s activity.
Protein structures have traditionally been determined by experimental methods such as x-ray crystallography and electron microscopy. But researchers have long wished to be able to predict a structure purely from its sequence—in other words, to understand and predict the process of protein folding.
For many years, computational methods such as molecular dynamics simulations struggled with the complexity of that problem. But AlphaFold bypassed the need to simulate the folding process. Instead, the algorithm could be trained to recognize correlations between sequence and structure in known protein structures and then to generalize those relationships to predict unknown structures.
After the first version of the algorithm was unveiled in 2018, the DeepMind researchers made improvements [1] that, four years later, enabled them to predict structures for all 200 million known protein sequences across all domains of life from bacteria to humans [2]. The quality of the predictions varies—the algorithm assigns a confidence score for each—but many are very close to the structures determined by crystallography.
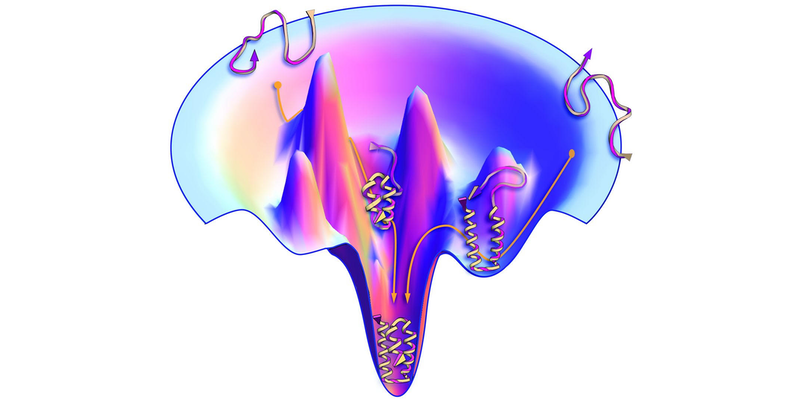
The protein-folding problem has long been of interest to statistical physicists because it exemplifies a general problem: how a complex system finds its lowest-energy, most-stable state. A protein molecule’s amino-acid chain can be folded up in a very large number of ways, and each folded configuration along the pathway to the final structure can be assigned an energy. This “energy landscape” has many local minima, and the puzzle is how the protein-folding process in nature finds its way to the global minimum without getting stuck in the “wrong” structure. That same problem of locating the global minimum in a complex energy landscape is encountered in various other physical systems, such as the magnetic materials called spin glasses.
Although AlphaFold sidesteps the difficulty of deducing the path of the folding chain through the energy landscape, some researchers would like to know if the algorithm nonetheless develops a representation—a kind of “intuition”—of the landscape from the training data. For a given sequence, the algorithm relies heavily on so-called coevolutionary data in the training set—structures for similar sequences with a few of the amino acids swapped out. These similar sequences could be interpreted as providing a “feel” for the relevant region of the energy landscape.
To test this idea, researchers recently trained AlphaFold using artificial coevolutionary data created with structure-prediction software that minimizes the total energy based on the interaction energies of all the amino acids. They concluded that the algorithm really does seem to sense the underlying energy landscape (see Viewpoint: Machine-Learning Model Reveals Protein-Folding Physics).
Its reliance on coevolutionary data is one of AlphaFold’s current limitations, however, according to theoretical chemist Peter Wolynes of Rice University in Texas, because it results in the algorithm too confidently insisting on a single structure. The algorithm may therefore struggle with proteins that change their structures as they carry out their biological functions and thus have more than one stable shape. And even when AlphaFold does successfully identify the shapes of fold-switching proteins, it seems to rely more on memorizing structures from its training data than on figuring them out from a deep representation of the energy landscape, according to another recent study [3].
Because of such provisos, AlphaFold, like all AI research tools, needs guidance from human experts. “Thoughtful use of machine learning has much to offer science if combined with human judgment,” says Wolynes.
–Philip Ball
Philip Ball is a freelance science writer in London. His latest book is How Life Works (Picador, 2024).
References
- J. Jumper et al., “Highly accurate protein structure prediction with AlphaFold,” Nature 596, 583 (2021).
- E. Callaway, “‘The entire protein universe’: AI predicts shape of nearly every known protein,” Nature 608, 15 (2022).
- D. Chakravarty et al., “AlphaFold predictions of fold-switched conformations are driven by structure memorization,” Nat. Commun. 15, 7296 (2024).