Machine-Learning Model Reveals Protein-Folding Physics
Proteins control every cell-level aspect of life, from immunity to brain activity. They are encoded by long sequences of compounds called amino acids that fold into large, complex 3D structures. Computational algorithms can model the physical amino-acid interactions that drive this folding [1]. But determining the resulting protein structures has remained challenging. In a recent breakthrough, a machine-learning model called AlphaFold [2] predicted the 3D structure of proteins from their amino-acid sequences. Now James Roney and Sergey Ovchinnikov of Harvard University have shown that AlphaFold has learned how to predict protein folding in a way that reflects the underlying physical amino-acid interactions [3]. This finding suggests that machine learning could guide the understanding of physical processes too complex to be accurately modeled from first principles.
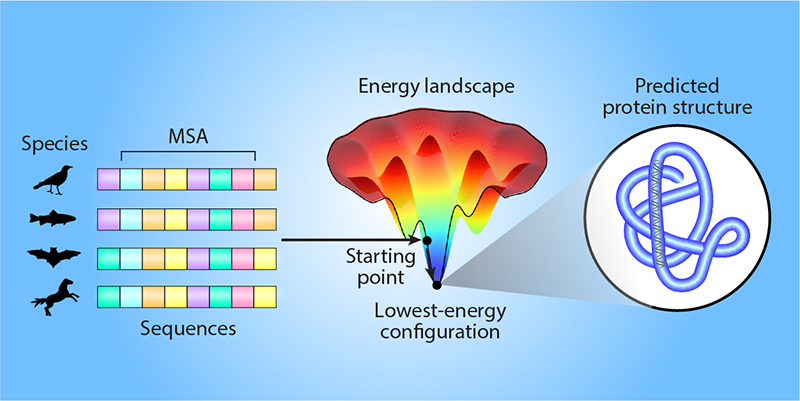
Predicting the 3D structure of a specific protein is difficult because of the sheer number of ways in which the amino-acid sequence could fold. AlphaFold can start its computational search for the likely structure from a template (a known structure for similar proteins). Alternatively, and more commonly, AlphaFold can use information about the biological evolution of amino-acid sequences in the same protein family (proteins with similar functions that likely have comparable folds). This information is helpful because consistent correlated evolutionary changes in pairs of amino acids can indicate that these amino acids directly interact, even though they may be far in sequence from each other [4, 5]. Such information can be extracted from the multiple sequence alignments (MSAs) of protein families, determined from, for example, evolutionary variations of sequences across different biological species. However, this reliance on MSAs is restrictive because such evolutionary knowledge is not available for all proteins.
Roney and Ovchinnikov hypothesize that the protein-folding model inferred by AlphaFold goes beyond MSA information (Fig. 1). They argue that this information only guides AlphaFold to a specific starting point on the energy landscape of protein folding—a map between the different 3D configurations of a given amino-acid sequence and their associated energies. Then, AlphaFold uses an “effective energy potential” that it has learned to locally search the energy landscape for the lowest-energy configuration, which corresponds to the likely 3D protein structure. The team tested this hypothesis using several computational experiments, in which AlphaFold ranked the quality of candidate protein structures that had previously been computationally predicted for different amino-acid sequences. AlphaFold was able to accurately rank the quality of the candidate structures in a way that was consistent with physical protein-folding models and that did not rely on any evolutionary information. These results indicate that the researchers’ hypothesis is likely correct.
The energy potential for protein folding learned by AlphaFold could open paths to exciting applications. Roney and Ovchinnikov suggest using the potential more broadly, for example, to explore how to fold de novo proteins (those designed in the lab from scratch) that lack MSA information or templates. Further investigation of the effective physical model uncovered by AlphaFold could reveal how amino-acid sequences spontaneously fold into their 3D structures inside cells. Moreover, the energy potential could be used to design sequences that fold into desired protein structures. Indeed, related machine-learning methods [6, 7] have already shown promise in designing viable sequences with desired folds for de novo proteins. It remains to be seen whether these methods implicitly leverage information about the underlying physics in their protein design process.
In the past few years, machine learning has revolutionized many aspects of protein science. AlphaFold is a success story in protein folding. Other kinds of machine-learning models that were originally designed to characterize the distributions of words in human languages have determined functional motifs in amino-acid sequences [8, 9]. The fact that AlphaFold has learned an energy potential, without specifically being trained to do so, indicates that efficient machine-learning algorithms can uncover key information about the physical interactions within molecules. Consistently, other types of efficient machine-learning algorithms that are trained to characterize protein structure-to-function maps have implicitly uncovered physical models for interatomic interactions [10].
The success of Roney and Ovchinnikov in constructing an energy potential from the predictions of AlphaFold reinforces the need to develop machine-learning models that are amenable to physical interpretation. This feature could also lead to more generalizability: if the implicitly learned physical laws could be made explicit, they could be used to solve problems beyond what the machine-learning models were originally trained to do. For protein science, it is certainly desirable for the next generation of machine-learning models to be physically interpretable.
References
- R. F. Alford et al., “The Rosetta all-atom energy function for macromolecular modeling and design,” J. Chem. Theory Comput. 13, 3031 (2017).
- J. Jumper et al., “Highly accurate protein structure prediction with AlphaFold,” Nature 596, 583 (2021).
- J. P. Roney and S. Ovchinnikov, “State-of-the-art estimation of protein model accuracy using AlphaFold,” Phys. Rev. Lett. 129, 238101 (2022).
- F. Morcos et al., “Direct-coupling analysis of residue coevolution captures native contacts across many protein families,” Proc. Natl. Acad. Sci. U.S.A. 108, E1293 (2011).
- D. S. Marks et al., “Protein 3D structure computed from evolutionary sequence variation,” PLoS ONE 6, e28766 (2011).
- J. Dauparas et al., “Robust deep learning–based protein sequence design using ProteinMPNN,” Science 378, 49 (2022).
- C. Hsu et al., “Learning inverse folding from millions of predicted structures,” bioRxiv (2022).
- A. Madani et al., “ProGen: Language modeling for protein generation,” bioRxiv (2020).
- A. Rives et al., “Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences,” Proc. Natl. Acad. Sci. U.S.A. 118 (2021).
- M. N. Pun et al., “Learning the shape of protein micro-environments with a holographic convolutional neural network,” arXiv:2211.02936.