How to Pop a Microscopic Cork
Researchers have designed a tiny structure that could help deliver drugs inside the body [1]. The theoretical and computational work required machine learning to optimize the parameters for the structure, which could stick to a closed shell containing a small molecule and cause the shell to open. The results demonstrate the potential for machine learning to assist in the development of artificial systems that can perform complex biomolecular processes.
Researchers are developing artificial molecular-scale structures that could perform functions such as drug delivery or gene editing. Creating such artificial systems, however, usually entails a frustrating tradeoff. If the components are simple enough to be computationally tractable, they are unlikely to yield complex interactions. But if the components are too complex, they become harder to combine and coordinate. Machine learning can reduce the computational cost of designing useful artificial systems, according to graduate student Ryan Krueger of Harvard University.
Krueger, working with Ella King of New York University and Michael Brenner of Harvard, focused his computational work on a shell made from a cluster of 12 spherical particles, each of which had sticky patches on its surface. The patches were positioned so that the spheres would form an icosahedral cage. A “cargo” molecule inside the cage could be released at the right time by removing one of the spheres. The researchers assigned the sphere-removal job to what they call a spider—a rigid pyramidal structure that would fit on top of the 12-sphere cluster. The spider consisted of a pentagonal ring of particles forming the base of the pyramid and a single head particle at the pyramid’s apex.
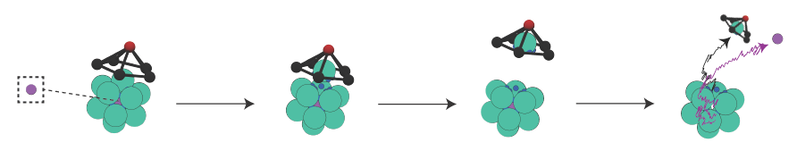
In the simulations, the icosahedral cluster was preassembled and fixed in position, whereas the spider was free to land anywhere on the cluster and interact with it. The particles of the spider’s base were neither attracted to nor repelled by the cluster. However, the head particle was attracted to the patches of the cluster particles by a force of adjustable range and strength. The spider’s dimensions and the radii of the head and base particles were also adjustable.
Krueger and his collaborators based their simulations on a standard computational technique called molecular dynamics, which calculates the motion of each particle in response to the interparticle forces. However, the researchers also wanted to determine which spider parameters would lead to the desired outcome: plucking away one sphere without destroying what remained of the cluster. Meeting that goal entailed not only calculating the interparticle forces but also optimizing a set of control parameters—the spider’s dimensions and the head particle’s attractiveness. With conventional molecular dynamics, these two simultaneous tasks would require too much computational power to accomplish in a reasonable time.
However, with the help of machine learning, the simulations succeeded in defining a spider that could meet its assigned task. In another set of runs, the researchers allowed the previously rigid spider to flex. This new degree of freedom—conformational entropy—served as an additional control parameter. Once optimized along with the others, it reduced the energy needed to extract a cluster particle.
Materials scientist Yang Jiao of Arizona State University is impressed by the work. He notes that previous work tended to set an equilibrium structure as the final target. By contrast, Krueger and his collaborators targeted a dynamic reaction. “This is a general computational framework, which, in principle, can be adopted to design a wide range of other complex reaction dynamics and pathways,” he says.
Implicit in the optimization framework is the ability to adjust control parameters almost continuously. Krueger notes that other systems for which such fine tuning is possible are self-assembling DNA-coated colloidal particles, which have potential applications in photonics and biomedical devices. In that case, Krueger envisions optimizing the DNA sequences in order to achieve specific behaviors.
–Charles Day
Charles Day is a Senior Editor for Physics Magazine.
References
- R. K. Krueger et al., “Tuning colloidal reactions,” Phys. Rev. Lett. 133, 228201 (2024).