How to Compare Proteins
Proteins play a central role in nearly every biological process, and they often change shape as they function. Over the past decade, a research team has developed a method of analysis that can help make sense of the available atomic-scale structural data and reveal the key physical distortions that underlie protein functions. Now the team has shown that the technique provides a consistent way of comparing proteins from different species, demonstrating similar structural changes in many of them [1]. The researchers believe that the technique will help biologists better understand the cross-species variations among proteins.
Proteins are linear chains of amino acids that fold up into specific three-dimensional shapes. Although there are lots of atomic-scale data on the structural changes that protein molecules execute as they function, researchers have had few quantitative methods to extract insights from these data, says biophysicist Pablo Sartori of the Gulbenkian Institute of Science in Portugal. One challenge, he says, is the arbitrary choice one makes when comparing two similar protein structures, such as the structures of a protein in two different conformations. “If you align region A of the protein, then region B shows displacement. If you align region B, then region A shows displacement. If you align the average, then both are displaced a bit.” Another problem is that the relative displacement is often not the quantity that best reflects the structural changes associated with protein function. If you imagine two connected rods that can close together like chopsticks, “when the hinge deforms, the rods move a lot, but the deformation actually occurs on the hinge, which moves very little,” Sartori says.
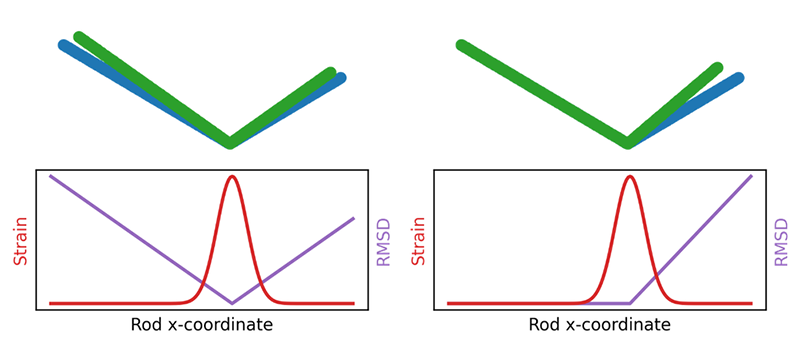
Eight years ago, to address this problem, biologist Stanislas Leibler of Rockefeller University in New York and his colleagues—inspired by work 20 years earlier [2]—adapted a method from materials physics known as finite strain analysis [3]. This mathematical technique compares an object’s initial state with its distorted state and gives a map that describes how the distortion varies across all parts of the object, with no alignment required. At any point, the object may be stretched, compressed, or rotated.
This measure of strain worked for individual proteins, and now Sartori and Leibler have used it to address the problem of comparing proteins across species. Researchers typically compare the proteins’ amino-acid sequences and look for identical, or “conserved,” regions, which often signify locations in the protein that are essential for its function. Sartori and Leibler suggest that a more direct way to find similar functioning regions is to compare the proteins’ strain maps.
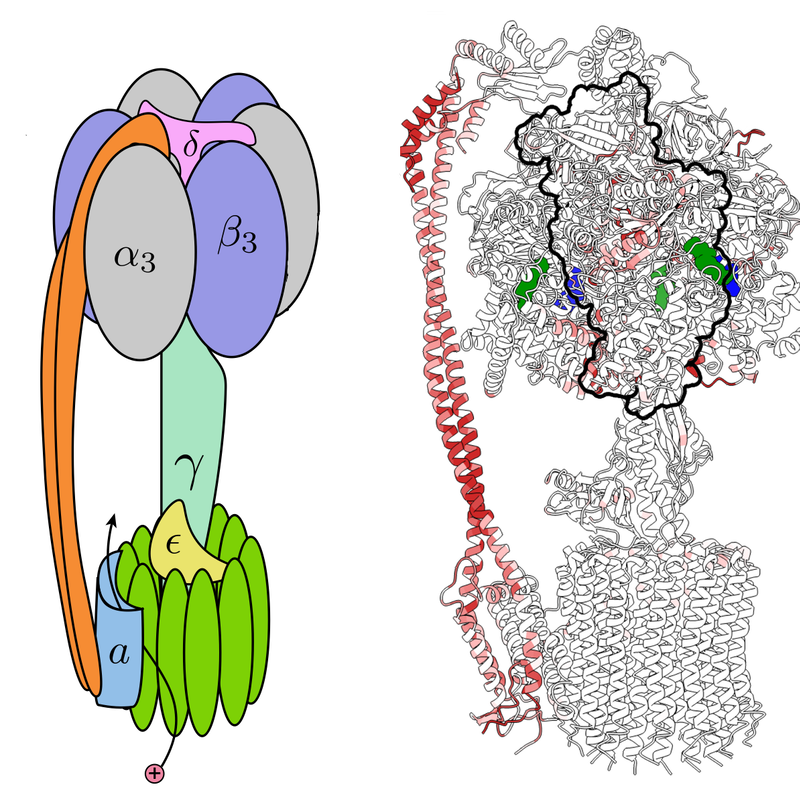
The duo focused on ATP synthase, a protein complex that synthesizes adenosine triphosphate (ATP), a molecule that stores energy. A flow of protons through the complex drives a relative rotation of two large subunits called Fo and F1, which are connected along their rotation axis by a “central stalk” and also linked by a so-called peripheral stalk. The rotation causes three smaller subunits 𝛽1, 𝛽2, and 𝛽3 to generate three new ATP molecules with each rotation cycle.
Using available data on ATP synthase derived from spinach leaves, Sartori and Leibler computed a strain map for the complex. The analysis shows large distortions—essentially, a local stretching—in several locations, including the middle region of the peripheral stalk and three regions where the tip of the central stalk interacts with 𝛽1, 𝛽2, and 𝛽3. While consistent with theoretical expectations, these distortions were not clear from previous analyses.
Next, Sartori and Leibler studied the patterns of strain in several ATP synthase complexes derived from other species, including Bacillus PS3, a common soil bacterium; Bos taurus, domesticated cows; and Saccharomyces cerevisiae, a common yeast. In general, the patterns of strain in these synthases were different in many regions, but they showed very similar strain in the regions of the key subunits 𝛽1, 𝛽2, and 𝛽3. Most significantly, the patterns of strain near these key subunits were more similar than the proteins’ amino-acid sequences. Sartori says that, in addition to the sequence, strain seems to be a useful marker of protein function.
To biophysicist Joe Howard of Yale University, this finding suggests an important change in perspective on how to identify the important structural changes underlying protein function. There are many kinds of motions in a protein, but not all of them are essential to its function. Howard says that according to the new work, strain is the key to finding the important molecular deformations, as it is maximized at those locations. “Consistent with this idea, they find that the [most highly] strained regions of ATP synthases are highly evolutionarily conserved among organisms,” he says.
Correction (12 March 2024): A paraphrase of Sartori’s comments has been updated to more accurately reflect his thoughts.
–Mark Buchanan
Mark Buchanan is a freelance science writer who splits his time between Abergavenny, UK, and Notre Dame de Courson, France.
References
- P. Sartori and S. Leibler, “Evolutionary conservation of mechanical strain distributions in functional transitions of protein structures,” Phys. Rev. X 14, 011042 (2024).
- T. Yamato, “Strain tensor field in proteins,” J. Mol. Graphics 14, 105 (1996).
- M. R. Mitchell et al., “Strain analysis of protein structures and low dimensionality of mechanical allosteric couplings,” Proc. Natl. Acad. Sci. U.S.A. 113 (2016).