Ionization Delays That Stand Out
The photoelectric effect—the emission of an electron upon absorption of a single photon—is the most basic manifestation of the interaction between light and matter. First explained by Einstein in 1905, it demonstrated the discrete nature of light, posing a crucial step in the development of quantum mechanics. But what happens exactly as an electron is ionized by a photon? How fast and by what mechanisms does it leave the trapping potential of the atoms or molecules to which it was bound? Physicist Eugene Wigner developed the theory to describe the process in the 1950s [1]. But until recently, no experimental probe was fast enough to catch the dynamics of electrons. The advent of attosecond (1 as =10−18 s) metrology is finally allowing researchers to probe electron ionization dynamics in real time.
Attosecond experiments have revealed that the photoionization of electrons occurs with measurable delays in atomic gases [2, 3] and solids [4, 5]. However, compared to atoms (or solids made of only one atomic species), measuring photoionization delays in molecules is more challenging. This is because molecules have both a higher density of states and an anisotropic electrostatic potential arising from the molecular geometry. These two factors complicate the interpretation of ionization measurements. Now, Martin Huppert, at the Swiss Federal Institute of Technology (ETH) in Zurich, and colleagues [6] have overcome these challenges, obtaining measurements of attosecond ionization delays in two molecules for a range of photon energies. They find that ionization delays in a water molecule are as short as those of the simplest atom (hydrogen), while the delays for another molecule ( N2O) can be much larger for photon energies corresponding to characteristic molecular resonances.
The length of ionization delays provides important information on the electronic structure of matter. These delays arise from the interactions of electrons with their environment, typically in the form of a potential representing the molecule’s electronic structure. Measuring such delays can thus shed light on the details of the potentials in which electrons move, which can help us develop and validate theoretical models for molecules. Such advances could ultimately open the door to controlling matter at its most fundamental level, enabling scientists to design molecules with desired electronic behavior.
Wigner’s theory introduced an analytic expression for the time shift in the outgoing electron wave packet due to the presence of a potential [1]. But the experimental characterization of such delays had to wait over half a century. In 2010, an influential experiment [2] used an attosecond extreme-ultraviolet (XUV) pulse to liberate electrons from the 2p and 2s orbitals of neon, and a second short infrared (IR) pulse to capture timing information on the departing electron. These measurements revealed that the 2p electrons are emitted with about a 20-as delay compared to the 2s electrons.
However, the extension of such measurements to molecules is challenging. The spectral lines of molecular spectra tend to be congested. This makes it extremely difficult to determine the initial electronic states excited by an attosecond XUV pulse, which contains a very broad range of frequencies. Additionally, in molecules, time delays can depend considerably on photoelectron energy, direction of emission, and the relative angle between the molecular axis and the polarization of the XUV pulse, further complicating the interpretation of experiments.
In atoms, the spherical symmetry of the potential considerably simplifies the interpretation of experiments. Hence, the photoelectron wave packet corresponding to ionization from each orbital (in the case of neon, 2s and 2p) can be described by just two “partial-wave” channels—two coherent quantum wave packets corresponding to the final angular momenta allowed by the selection rules. In principle, a similar description could be applied to molecules. However, many partial-wave channels are required to describe the more complex wave packet extending into the continuum around a molecule. Additionally, the angular interference between many partial waves, for a given electron energy, gives rise to a rich angular structure and to a strong energy dependence of the photoionization cross section and time delays [7]. Huppert et al. [6] managed to overcome these obstacles by applying a variety of techniques that minimize the signal-to-noise ratio of their experiments and by interpreting their data using scattering models [7, 8] that can lend themselves to a partial-wave analysis for situations more complex than atoms. This allowed them to gain a clear picture of how photoionization delays depended on energy.
The authors’ scheme [6] combined an XUV attosecond pulse train (APT), made of high-order odd harmonics from an 800-nm-wavelength laser, with a second IR pulse that was synchronized with the XUV beam. The XUV radiation ionizes the electrons, while the IR pulse is used as a temporal probe of the outgoing photoelectron [9]. Timing information is derived by measuring, as a function of the delay between the two beams, interference effects between different XUV and IR two-photon transitions leading to the same final electron energy. (This method was previously used to measure atomic ionization delays in argon [3] and solids [5]). To isolate different energies, the authors used three types of metal foils, which selectively filter the 11th–15th, 15th–21st, and 23rd–27th harmonics contained in the APT. Thanks to this strategy, they easily identified signals coming from different electronic states within the molecules. They obtained ionization delays between the two lowest-lying electronic states of N2O and H2O over a range of photon energies within the bandwidth of the 11th–27th harmonics of the XUV spectrum.
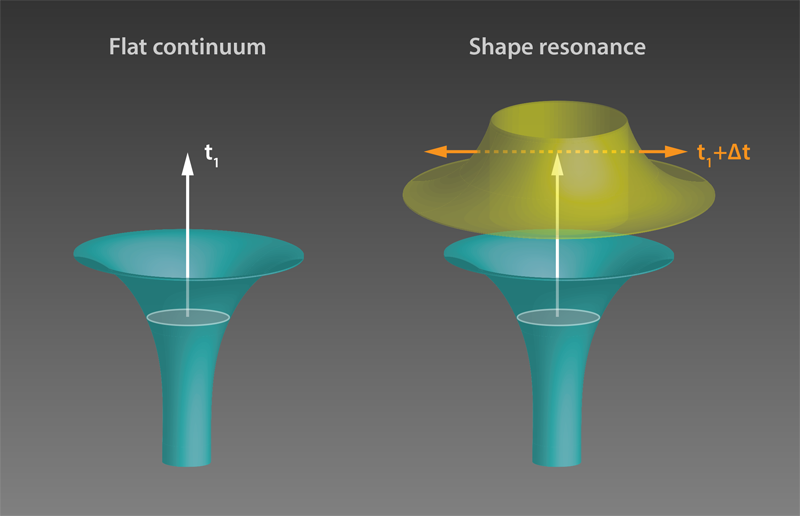
The measured delays in H2O molecules were shorter than 50 as and decreased monotonically with energy. This energy dependence can be understood by considering that a higher energy electron can more easily climb the atomic potential and is thus slowed down less. On the other hand, time delays in N2O showed a more varied behavior, reaching surprisingly large values of up to 160 as at certain photon energies. To understand these results, the authors used sophisticated theoretical tools based on scattering theory [7–9]. The observed large delays in N2O were attributed to a “shape resonance” (see Fig 1). Shape resonances occur when the wave packet of the departing electron matches the shape of the trapping potential, which is determined by the molecular potential and by a centrifugal contribution from the electron’s angular momentum. Under this condition, photoionization is predicted to slow down. The authors’ findings are confirmed by the existence of a well-known shape resonance associated with an electronic state of the molecule ( Ã+) .
The work by Huppert et al. [6] is an important milestone in the investigation of molecular photoionization dynamics. Moreover, it complements recent studies on solid surfaces [5, 10], in which ionization delays can also be attributed to resonance effects involving excited electronic states. Combined with knowledge of molecular orientation, these techniques could provide new insights into previously inaccessible properties of a molecule’s electronic structure including electronic correlations and the localization of electronic wave packets within the molecules.
This research is published in Physical Review Letters.
References
- E. P. Wigner, “Lower Limit for the Energy Derivative of the Scattering Phase Shift,” Phys. Rev. 98, 145 (1955).
- M. Schultze et al., “Delay in Photoemission,” Science 328, 1658 (2010).
- K. Klünder et al., “Probing Single-Photon Ionization on the Attosecond Time Scale,” Phys. Rev. Lett. 106, 143002 (2011).
- A. L. Cavalieri et al., “Attosecond Spectroscopy in Condensed Matter,” Nature 449, 1029 (2007).
- R. Locher, L. Castiglioni, M. Lucchini, M. Greif, L. Gallmann, J. Osterwalder, M. Hengsberger, and U. Keller, “Energy-Dependent Photoemission Delays From Noble Metal Surfaces by Attosecond Interferometry,” Optica 2, 405 (2015).
- M. Huppert et al., “Attosecond Delays in Molecular Photoionization,” Phys. Rev. Lett. 117, 093001 (2016).
- P. Hockett, E. Frumker, D. M. Villeneuve, and P. B. Corkum, “Time Delay in Molecular Photoionization,” J. Phys. B 49, 095602 (2016).
- J. M. Dahlström, D. Guénot, K. Klünder, M. Gisselbrecht, J. Mauritsson, A. L’Huillier, A. Maquet, and R. Taïeb, “Theory of Attosecond Delays in Laser-Assisted Photoionization,” Chem. Phys. 414, 53 (2013).
- The setup in Ref. [6] offers higher spectral resolution due to the periodicity of the pulses because the XUV spectrum is made up of a series of discrete odd harmonics. (An isolated attosecond pulse as used in Refs. [2,4] would instead have a broad continuous spectrum.).
- Z. Tao, C. Chen, T. Szilvasi, M. Keller, M. Mavrikakis, H. Kapteyn, and M. Murnane, “Direct Time-Domain Observation of Attosecond Final-State Lifetimes in Photoemission From Solids,” Science 353, 62 (2016).