Timing Molecular Motion with an Optical Stopwatch

The ionization of a molecule by light sounds simple—the light comes in, electrons get kicked out. But ask about the details of this process—such as how the molecule reconfigures in response—and the theoretical picture is murky for all but a few textbook cases. A set of techniques for controlling and imaging molecules has now reached a new level of temporal and spatial resolution to provide some clarity. Markus Kitzler-Zeiler of Vienna University of Technology (TU Vienna) and colleagues imaged the wave function of a simple molecule as it responded to the loss of an electron [1]. Combining two state-of-the-art techniques, they produced a “movie” of the molecule that captured the movements of the molecule’s protons in 1 picometer ( 10−12 m) increments—a small fraction of the width of an atom—and with 70 attosecond ( 7×10−17s) resolution. The approach allows a test of ionization models in H2 and may lead to a better understanding of chemical processes in more complex molecules.
The researchers focus on the simple motion of protons in singly ionized molecular hydrogen ( H+2). With a simple structure of just two protons bound together by an electron, H+2is for molecular physics what the fruit fly is for genetics. In an experiment, the team prepares an ensemble of cold H2 molecules by injecting gas through a supersonic nozzle and into a vacuum chamber. When an H2 molecule is intercepted by a sufficiently strong laser pulse, the laser’s electric field removes one electron, which sets the now weakly bound protons into an oscillatory motion mediated by the remaining electron. This oscillation would continue indefinitely, were it not for the still-present laser field, which causes the second electron to be ejected at some random time ( Δt) after the first. With nothing left to keep the protons together, their mutual repulsion pushes them apart in a “Coulomb explosion” [2].
The time between the first and second ionization is anywhere between 0 and about 5 fs, and it is the increasing separation between the two protons during this interval that the researchers “image” by combining two techniques (Fig. 1). In the first technique, they use their laser pulse as a sort of “stopwatch” that starts when the first ejected electron is detected and stops when the second ejected electron is detected. In fact, the pulse is just two cycles of elliptically polarized, 750-nm light, and its rotating electrical field vector serves as the ticking hand of a watch that makes a full rotation every 2.5 fs. The researchers can infer the “ticks” that dislodge the first and second electrons by measuring the direction that they escape—a technique known as angular streaking [3].
The second experimental ingredient is a “picoscope,” which extracts the distance between the protons when the molecule explodes by measuring their kinetic energy. When the laser pulse strips off the second electron, each proton feels a 1∕r repulsive Coulomb potential. Each proton then “slides down” this potential by moving further away from its partner, gaining kinetic energy by an amount that depends on its initial distance from the other proton. The team measured this kinetic energy at the time of the explosion and compared it to the known potential, achieving an effective “magnifying glass” that sees the proton separation when the molecule breaks apart. Much like particle physicists, the researchers measured the kinetic energy of the protons by following their trajectories in electric and magnetic fields [4].
Each timed Coulomb explosion serves as a mini experiment. After roughly 10 million such experiments, the researchers reconstructed the proton-proton distance at several time intervals after the first ionization (Fig. 2). When viewed together, these reconstructions essentially provide a time-lapsed, picometer-resolution image of the molecular wave function—both the separation between the protons and the proton’s quantum-mechanically smeared out position. Although this demonstration is primarily a proof-of-principle, the researchers were also able to use their precise mapping of the wave function to test and rule out two ionization scenarios for H2, known as shake-up and resonant multiphoton electronic excitations.
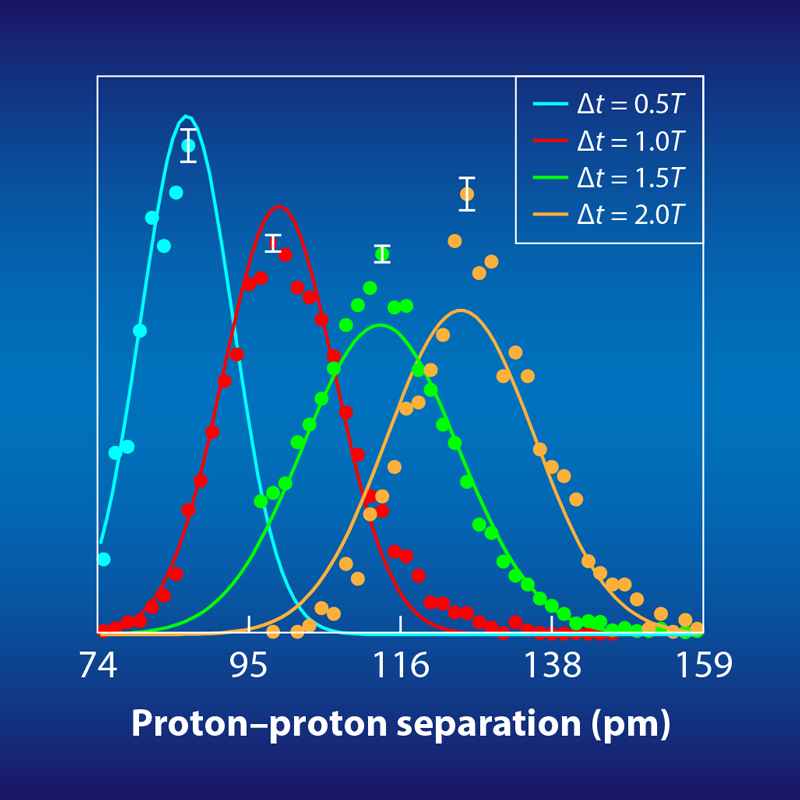
This is the first time an ionized molecule has been viewed with such high spatial and temporal resolution at once. Having validated this approach with the molecular fruit fly, the next step would be to apply it to more complicated and less studied species, particularly those that are of interest to chemistry, molecular biology, and medicine. Applying the picoscope to more complicated molecules will require knowledge of more complicated molecular potential surfaces. However, Kitzler-Zeiler and colleagues contend this is not a huge problem since such surfaces can be readily calculated, even if the molecular dynamics preceding the Coulomb explosion cannot. From my view, an interesting avenue to explore would be the process of quantum tunneling. When an electron is removed from the H+2 ion by the strong laser field, it spends time “under the barrier” of the ion’s potential [5]. But the length of time and the behavior of the ion during that time are fascinating unknowns that the new technique might potentially explore.
The art of molecular “movie making” has advanced significantly since the 1980s and 1990s, when Ahmed Zewail and his colleagues first used femtosecond laser pulses to make movies of molecular oscillations and chemical reactions—an accomplishment that was recognized with the 1999 Nobel Prize in Chemistry. Zewail adopted a “pump-probe” technique, where one pulse (the pump) excites a collection of molecules and a subsequent weaker pulse (the probe) samples changes in the molecular state. The inherent time resolution of his approach was on the order of the fs duration of the “envelope” of the laser pulse—the smooth rise and fall in intensity that modulates the much more rapidly varying optical cycles. By contrast, Kitzler-Zeiler and colleagues achieved a sub-fs probe by tapping into the “carrier” electric field, which rotates with the actual cycles of elliptically polarized light [6]. A further improvement might come from using available techniques to stabilize and exploit the relative phase between the carrier and the envelope [7]. This capability would allow the determination of not only the relative time between two ionization events, Δt, but also the absolute timing of the events. Nevertheless, with the relative timekeeping already in hand, the researchers have obtained an unprecedented view of both the position of a molecular wavepacket and its shape.
This research is published in Physical Review Letters.
References
- V. Hanus et al., “Subfemtosecond tracing of molecular dynamics during strong-field interaction,” Phys. Rev. Lett. 123, 263201 (2019).
- H. Stapelfeldt et al., “Time-resolved Coulomb explosion imaging: A method to measure structure and dynamics of molecular nuclear wave packets,” Phys. Rev. A 58, 426 (1998); H. Niikura et al., “Probing molecular dynamics with attosecond resolution using correlated wave packet pairs,” Nature 421, 826 (2003).
- J. Wu et al., “Probing the tunnelling site of electrons in strong field enhanced ionization of molecules,” Nat. Commun. 3, 1113 (2012).
- R. Dörner et al., “Cold target recoil ion momentum spectroscopy: A ‘momentum microscope’ to view atomic collision dynamics,” Phys. Rep. 330, 95 (2000).
- L. V. Keldysh, “Ionization in the field of a strong electromagnetic wave,” Sov. Phys. JETP 20, 1307 (1965); P. Eckle et al., “Attosecond ionization and tunneling delay time measurements in helium,” Science 322, 1525 (2008).
- P. B. Corkum and F. Krausz, “Attosecond science,” Nat. Phys. 3, 381 (2007); F. Krausz and M. Ivanov, “Attosecond physics,” Rev. Mod. Phys. 81, 163 (2009); B. Wolter et al., “Ultrafast electron diffraction imaging of bond breaking in di-ionized acetylene,” Science 354, 308 (2016).
- A. Baltuška et al., “Attosecond control of electronic processes by intense light fields,” Nature 421, 611 (2003).