Showtime for Molecular Movies
The scientist Ahmed Zewail (1946–2016) was awarded the Nobel Prize in Chemistry in 1999 for his contributions to femtochemistry, the field that studies chemical changes on the time scales on which atoms move—femtoseconds (fs). Femtochemistry experiments follow the electronic or structural dynamics of a molecule by first exciting it with a femtosecond “pump” laser pulse, and then observing it using a delayed “probe” pulse [1]. Most femtochemistry experiments, however, probe molecules through indirect observables: They rely on the fact that the chemical dynamics are accompanied by changes in the molecule’s absorption spectrum. Two independent groups now use more direct approaches to the visualization of atomic motions, recording molecular “movies” by time-resolved x-ray diffraction [2] and time-resolved electron diffraction [3], respectively. Both experiments image, in real time, the changes of the distance separating the two atoms in a vibrating iodine ( I2) molecule. The results represent important progress in our ability to record ultrafast molecular movies with increasing spatial and temporal resolution without relying on complex models.
In conventional femtochemistry approaches, the evolution of a molecule cannot be extracted from an experiment without knowledge of the molecule’s potential energy surfaces. These surfaces describe the electronic states and their dependence on the molecular structure. However, this requirement severely limits the range of systems that can be studied, or the structural information that can be derived from experiments. A number of groups, including Zewail’s, thus moved to develop techniques that can directly probe molecular structural dynamics, such as time-resolved x-ray and electron scattering [4–6]. Such experiments provide a much more direct view of an evolving molecular structure, since internuclear distances and scattering patterns are directly related through a simple diffraction relation. In other words, they offer the promise of recording a molecular movie.
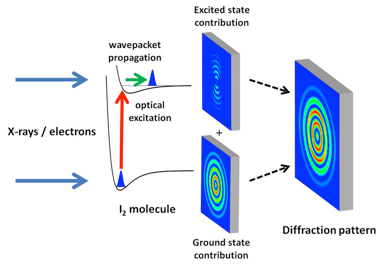
X-ray experiments received a major impetus with the development of x-ray free-electron lasers, like the Linac Coherent Light Source (LCLS), which opened in 2009 at SLAC National Accelerator Laboratory [7]. The brightness of x-ray free-electron lasers exceeds that of conventional x-ray sources by many orders of magnitude. At the same time, researchers reported great progress in the development of stable and intense electron pulses with increasingly shorter time durations [8]. Relying on such technological advances, the two new studies image the internuclear distance between the two atoms in I2 after the molecule has been excited from its electronic ground state to an electronically excited state by a visible femtosecond pulse (see Fig. 1). This impulsive electronic excitation creates a vibrational wave packet that moves within the excited molecule, modulating the internuclear distance.
James M. Glownia at SLAC National Accelerator Laboratory, California, and co-workers map internuclear distances using x-ray photons from the LCLS [2]. They demonstrate how x-ray scattering from photoexcited molecules can be considered a holographic experiment. In holography, a sample is imaged by measuring the coherent interference between a beam that interacted with the sample and a reference beam that did not. Similarly, the x-ray scattering signal in Glownia et al.’s experiment can be regarded as the mixing of two contributions [9]. The first comes from the fraction of molecules in the gas sample that were excited by the visible pump laser (an experiment can typically excite 1–10% of the molecules). These molecules are displaying a vibrational wave-packet motion and produce a weak x-ray scattering pattern that, by itself, is not measurable. However, this signal becomes measurable through its interference with the stronger x-ray scattering pattern from the more abundant ground-state molecules. By subtracting a ground-state-only scattering pattern (which they measure when the x-ray probe precedes the visible pump), the authors are able to isolate the ground-state and excited-state interference contribution to the signals. Through simple mathematical manipulations (Fourier transformations), the internuclear distance can be determined from the interference pattern formed by the ground-state and excited-state signals.
It is important to note that this data analysis does not rely on complex molecular modeling. As such, this approach represents a major advance compared to previous x-ray diffraction experiments [6]. By carrying out their measurement for a series of pump-probe time delays, the authors create a molecular movie, beautifully showing oscillations in the internuclear distance of the vibrational wave packet, with a period of a few hundred femtoseconds. The researchers achieve an exceptional combination of temporal (30 fs) and spatial (.3 Å) resolution. The movie also reveals other small contributions from photoinduced fragmentation of the molecule.
A similar scattering experiment was carried out by Jie Yang at the University of Nebraska, Lincoln, and colleagues [3], with the difference that the x rays are replaced by a beam of relativistic, 3.7 mega-electron-volt (MeV) electrons. The source, also located at SLAC National Accelerator Laboratory, generates electron pulses by shining a laser onto a metallic photocathode. The pulses are accelerated with a radio frequency field and focused onto the sample by a magnetic lens [9]. The use of 3.7-MeV electron energies, more than an order of magnitude larger than in most earlier electron diffraction experiments, allowed the researchers to overcome two problems that have previously limited the time resolution in gas-phase ultrafast electron diffraction experiments. The first is due to the fact that electrons do not move at the speed of light: in laser-pump–electron-probe experiments, this causes a mismatch between laser pulse and electron velocities that deteriorates the temporal resolution. The second is the mutual Coulomb repulsion between the electrons in the beam, which broadens the electron pulses. At 3.7 MeV, the electrons move almost at the speed of light and experience reduced Coulomb broadening. Consequently, Yang et al. reached a time resolution of 230 fs in their pump-probe experiment, sufficient for following the 400-fs-period of the vibrational motion of the iodine molecule. While the temporal resolution, which is limited by the electron pulse duration, could not reach that of Glownia and co-workers, the short electron wavelength led to an exquisite spatial resolution of 0.07 Å.
The authors consider it possible to achieve a sub-50-fs time resolution in the near future. This could be done with the planned inclusion of radio frequency schemes that further compress the electron bunch [7] or by increasing the pulse repetition rate while decreasing the number of electrons in each bunch (which reduces Coulomb repulsion effects). In principle, one may envision that a holographic approach similar to that of Glownia et al. might be applicable to the electron diffraction experiment as well. For now, Yang et al. treat the measured electron diffraction pattern as an incoherent sum of ground- and excited-state contributions, from which the excited-state-only contribution can be obtained by subtracting the ground-state one, which is readily obtained from a static measurement.
In conclusion, the two studies point to a bright future for recording a wide range of molecular movies on a time scale of a few tens of femtoseconds. Structural changes on even faster time scales may soon be measurable by using ever shorter electron or x-ray free-electron laser pulses [10], or by replacing the electrons from a photocathode source with electrons extracted from within the molecule under investigation, as done in laser-induced electron diffraction [11]. The novel methods promise to deliver new insights into fast, fundamental molecular processes that have been hard to observe. These include many mechanisms determining transitions between electronic states (such as internal conversion or intersystem crossing) that have important consequences for chemical dynamics. Similar experiments may also help researchers tackle biologically relevant problems such as photoisomerization and photoprotection.
This research is published in Physical Review Letters.
Correction (3 October 2016): An earlier version of the article incorrectly stated that the spatial resolution achieved by Glownia et al. was 3 Å. It is in fact 10 times lower, 0.3 Å.
Editor’s Note (11 August 2017): Kochise Bennett and colleagues have published a Comment in PRL that challenges the interpretation of the experiments by Glownia et al. For more information, see the Comment by Bennett et al. and the Reply by Glownia et al.
References
- A. H. Zewail, “Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond Using Ultrafast Lasers (Nobel Lecture),” Angew. Chem. Int. Ed. 39, 2586 (2000).
- J. M. Glownia, “Self-Referenced Coherent Diffraction X-ray Movie of Ångstrom- and Femtosecond-Scale Atomic Motion,” Phys. Rev. Lett. 117, 153003 (2016).
- J. Yang, “Diffractive Imaging of Coherent Nuclear Motion in Isolated Molecules,” Phys. Rev. Lett. 117, 153002 (2016).
- D. Shorokhov and A. H. Zewail, “Perspective: 4D Ultrafast Electron Microscopy—Evolutions and Revolutions,” J. Chem Phys. 144, 080901 (2016).
- G. Sciaini and R. J. D. Miller, “Femtosecond Electron Diffraction: Heralding the Era of Atomically Resolved Dynamics,” Rep. Prog. Phys. 74, 096101 (2011).
- P. Emma et al., “First Lasing and Operation of an Ångstrom-Wavelength Free-Electron Laser,” Nature Photonics 4, 641 (2010).
- T. van Oudheusden, P. L. E. M. Pasmans, S. B. van der Geer, M. J. de Loos, M. J. van der Wiel, and O. J. Luiten, “Compression of Subrelativistic Space-Charge-Dominated Electron Bunches for Single-Shot Femtosecond Electron Diffraction,” Phys. Rev. Lett. 105, 264801 (2010).
- M. J. J. Vrakking and T. Elsaesser, “X-Ray Photonics: X-rays Inspire Electron Movies,” Nature Photon. 6, 645 (2012).
- S. P. Weathersby et al., “Mega-Electron-Volt Ultrafast Electron Diffraction at SLAC National Accelerator Laboratory,” Rev. Sci. Instrum. 86, 073702 (2015).
- W. Helml et al., “Measuring the Temporal Structure of Few-Femtosecond Free-Electron Laser X-ray Pulses Directly in the Time Domain,” Nature Photon. 8, 950 (2014).
- C. I. Blaga, J. Xu, A. D. DiChiara, E. Sistrunk, K. Zhang, P. Agostini, T. A. Miller, L. F. DiMauro, and C. D. Lin, “Imaging Ultrafast Molecular Dynamics with Laser-Induced Electron Diffraction,” Nature 483, 194 (2012).