Selective Bond Breaking with Splat Chemistry

The ability to selectively excite and break specific bonds in molecules would open new vistas in synthetic chemistry, allowing the creation of compounds that are difficult to synthesize via conventional chemical techniques. However, decades of research have shown that, with a few exceptions [1], when energy is put into a specific molecular bond—with a laser, for example—it is quickly redistributed among many vibrations in the molecule long before a reaction may occur. In other words, attempting to selectively put energy into a bond usually leads to the same chemical reaction as heating the reactants on a hot plate. Surprisingly, Lukas Krumbein, at the Max Planck Institute for Solid State Research in Germany, and colleagues have now observed that a bond in a large molecule can be selectively broken by adding energy to the system in the simplest way possible—by colliding the molecule with a surface [2]. The result improves our understanding of the dynamics of large molecules and could offer novel ways to control their reaction products.
The idea that energy imparted during a collision can promote chemical reactions—a process sometimes called splat chemistry—has been around for decades. Researchers have shown, for instance, that the collision of argon atoms onto methane (CH4) molecules adsorbed on a surface can lead to the molecules’ dissociation [3]. In that experiment, the equivalence of the four C–H bonds means that the collision process is not bond specific. What is significant about the work by Krumbein and colleagues is that they demonstrate the cleavage of a particular bond.
The researchers use electrospray ionization and ion-beam deposition to accelerate a 73-atom molecule called Reichardt’s dye toward a copper surface. Using scanning tunneling microscopy to inspect the scene of the collision, they find that when the molecule collides with the surface at translational energies of 2–50 eV—larger than the energy associated with thermal excitation—it selectively “cracks” at a specific carbon–nitrogen (C–N) bond. Breaking this bond results in the molecule opening into a more spread-out configuration. In contrast, when the molecule is simply heated, a different C–N bond is broken, which “splits” the original molecule into two pieces. Krumbein and colleagues also observe this lower-energy splitting reaction in some collisions, but this reaction has a lower probability than the cracking reaction.
Key to the success of their experiment is the sheer size of the molecule involved. Previous studies of surface dynamics have focused on the reactions of small molecules, such as nitric oxide (NO) or CH4. The collision-induced excitation of single bonds in such small molecules is relatively ineffective. For example, when a molecule of NO collides with a surface, the efficient compression of the N–O bond would require a perfectly aligned, “head-on” collision geometry. With any other alignment, the molecule would behave much like an American football, hitting with its long axis at an angle to the surface [4, 5]. This off-axis geometry causes most of the translational energy to be converted into rotational energy, sending the molecule spinning away from the surface but without inducing any chemical change.
The situation is very different for Reichardt’s dye, named after the doctoral student who developed the molecule while searching for a compound that would change its color depending on the solvent. This large organic molecule consists of seven rings surrounding a central nitrogen atom. When a Reichardt’s dye molecule hits the surface, the collision doesn’t compress a single bond. Instead, the collision causes the entire molecule to undergo a large-scale distortion in a very short period of time—more like a prop-comedy rubber chicken hitting a wall than like a football.
Based on simulations of their experiment, Krumbein and colleagues explain that the fate of the molecule depends on its orientation when hitting the surface. The key difference between the collision geometries is how the large-scale distortion strains the three carbon atoms surrounding the central nitrogen atom (Fig. 1). Collisions that focus the molecule’s distortion on one particular carbon atom lead to splitting, with the molecule breaking apart into two fragments. Distortions focused on one of the other two (symmetry equivalent) carbon atoms lead to cracking, in which the molecule hinges open but remains in one piece. Other impact configurations produce no reaction, leaving all three bonds unbroken and the molecule intact (Fig. 2).
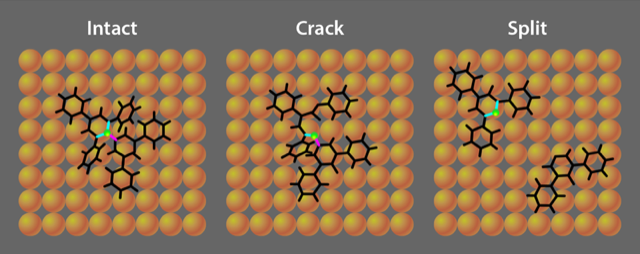
The experiments demonstrate that, as expected, the probability of a reaction is dependent on the initial translational energy of the molecule. Faster molecules split or crack with higher probability than slower molecules. Using simulations that account for the collision process, the researchers explain the experimental observation that the more common outcome is a cracking reaction, even though splitting is the thermally favored pathway. This is not contradictory, as the reaction rate is controlled both by the activation barrier and by the probability of attaining a suitable molecular configuration for the reaction. The simulations reveal that the collisions selectively strain the central nitrogen atom in a way that promotes C–N bond cleavage. In contrast, heating the molecule distributes energy randomly.
The results obtained by Krumbein and colleagues provide important new insight into the localization of energy in molecules: The large-scale distortion of the molecule focuses the energy on a single bond while simultaneously preventing the energy from rapidly delocalizing—at least for the picosecond or so that it takes for the molecule’s geometry to stabilize after the collision. These types of large-scale deformations are common in macromolecules under strain, such as polymers, proteins, and DNA. Accounting for the way that energy is concentrated on specific bonds within these molecules will help predict how they respond to such strain. Insights such as those provided by this work are also important for understanding mechanochemistry, the coupling between macroscopic strain and chemical reactivity. Mechanochemistry underlies important phenomena, such as stress-corrosion cracking and polymer degradation under shear. An atomistic understanding of mechanochemistry is still in its infancy, but Krumbein and colleagues’ experiment represents a considerable advance.
References
- R. L. Vander Wal and F. F. Crim, “Controlling the pathways in molecular decomposition: The vibrationally mediated photodissociation of water,” J. Chem. Phys. 93, 5331 (1989).
- L. Krumbein et al., “Fast molecular compression by a hyperthermal collision gives bond-selective mechanochemistry,” Phys. Rev. Lett. 126, 056001 (2021).
- J. D. Beckerle et al., “Collision-induced dissociative chemisorption of adsorbates: Chemistry with a hammer,” J. Chem. Phys. 86, 7236 (1987).
- G. M. McClelland et al., “Determination of internal-state distributions of surface scattered molecules: Incomplete rotational accommodation of NO on Ag(111),” Phys. Rev. Lett. 46, 831 (1981).
- A. W. Kleyn et al., “Rotational energy transfer in direct inelastic surface scattering: NO on Ag(111),” Phys. Rev. Lett. 47, 1169 (1981).