Deciphering Water’s Dielectric Constant
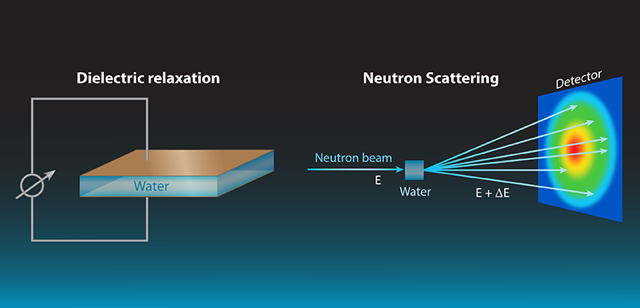
Water is an extremely complex liquid, made of networks of H2O molecules linked by hydrogen bonds that continuously form and break. Most of our knowledge of the liquid comes from spectroscopic studies—dielectric relaxation, Raman scattering, infrared absorption, and x-ray or neutron scattering—which use radiation or particles to probe different aspects of atomic and molecular motions. The combined information derived from all these techniques may provide a coherent picture of the liquid’s dynamics, shedding light on important questions about the nature of water. How do molecular phenomena, such as vibrations, rotations, diffusion, and the making and breaking of hydrogen bonds determine the macroscopic properties of water, including its large dielectric constant? What are the mechanisms by which the liquid relaxes when energy is deposited into it? What is the distance over which molecules of water can “feel” each other—the so-called correlation length?
Now, Arantxa Arbe and her colleagues [1] combine results from measurements of dielectric relaxation [2] and neutron scattering to deliver a detailed and unified picture of water dynamics at room temperature. Through dielectric relaxation experiments, the authors characterized the collective relaxation of electrical dipoles that determines the macroscopic dielectric response of water. Thanks to the atomic-scale sensitivity of neutron scattering measurements, they were then able to establish a relationship between molecular dynamics and the dielectric behavior of the liquid.
Measurements of dielectric relaxation [2] are one of the most widely used means to observe water dynamics. In its simplest version, the method applies an oscillating electric field to a capacitor containing the liquid, and measures, as a function of the field’s frequency, the phase difference between the current and voltage on the capacitor (see Fig. 1). From this measurement, one can extract the dielectric response of the liquid, which is related to the dynamics of the dipoles that were excited by the applied field.
Recently, experiments studied dielectric relaxation in pure water up to terahertz frequencies [2]. However, since the wavelength of the electromagnetic field used in this kind of experiment is of the order of centimeters, the technique probes the dynamics of the macroscopic dipole moment but can’t decipher the molecular details underlying the dielectric behavior.
By contrast, neutrons interact directly with atomic nuclei. A neutron scattering experiment (see Fig. 1) can measure both local atomic motions and larger-scale collective dynamics. Neutron scattering determines the so-called dynamic structure factor, or S(Q,𝜈), as a function of frequency 𝜈 and momentum transfer Q, which correspond, respectively, to the differences of energy and momentum between incoming and outgoing neutrons. S(Q,𝜈) is a correlation function between positions occupied by the same or different atoms at different times. Spatial information on molecular dynamics can be derived from the Q dependence, while temporal information can be accessed via the energy dependence.
The ability to selectively probe local and collective contributions to the dynamics stems from the properties of hydrogen nuclei. In neutron experiments, scattering by hydrogen atoms is incoherent—a type of direction-independent scattering that is related to the individual motions of hydrogen atoms but not to the liquid’s structure or collective dynamics. Deuterium (D) atoms, on the other hand, generate predominantly coherent scattering, which derives from collective motions. By comparing experiments on light (H2O) to heavy water (D2O) one can thus separate local and collective information.
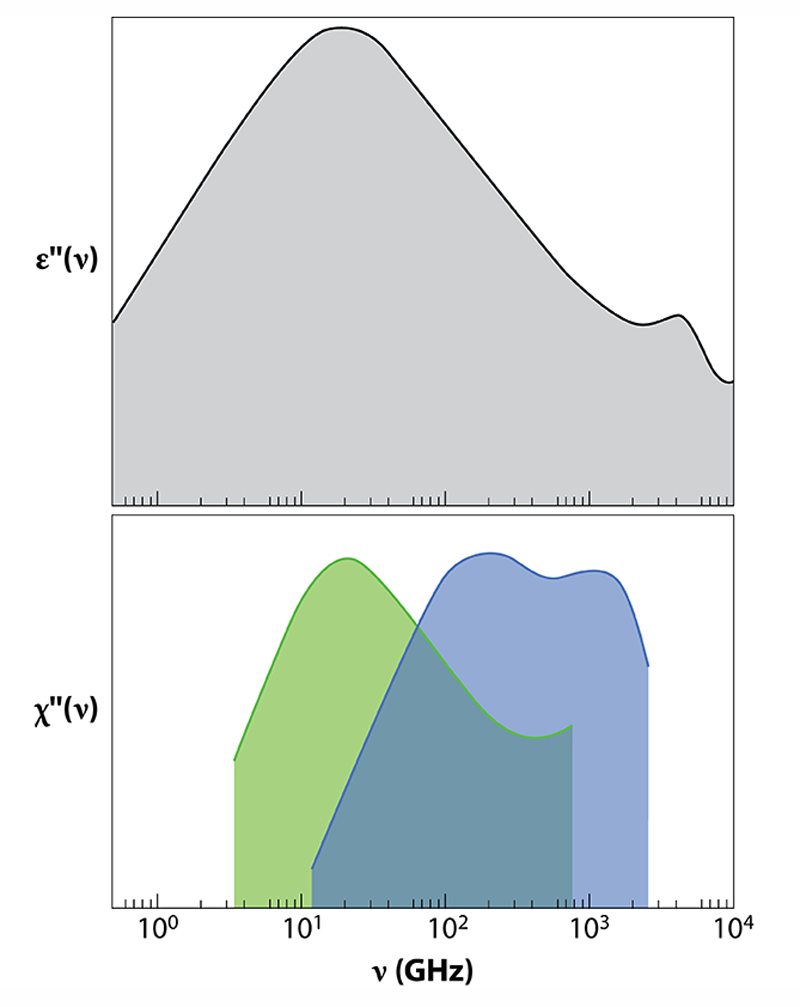
Studies of liquid water that combine dielectric relaxation and neutron scattering have previously been carried out, but they amounted to qualitative observations. Arbe et al. now provide the first quantitative study using the two techniques. The observables in the two experiments— 𝜀′′(𝜈) in dielectric relaxation and S(Q,𝜈) in neutron scattering—are directly related: S(Q,𝜈) can be connected to the dielectric susceptibility 𝜒′′(Q,𝜈)=(𝜈∕kBT)⋅S(Q,𝜈), where kB is the Boltzmann constant and T is the absolute temperature. 𝜒′′(Q,𝜈) is the equivalent of 𝜀′′(𝜈), the imaginary part of the dielectric constant measured in dielectric relaxation.
The main feature of dielectric relaxation measurements is a peak in 𝜀′′(𝜈) centered at 20 GHz. This is the well-known Debye peak, indicating that the collective relaxation of the macroscopic dipole moment occurs on a time scale of 8.3 ps (Fig. 2). Arbe and colleagues have now carried out coherent and incoherent neutron scattering experiments to shed new light on the molecular origin of this relaxation. The authors show that the same peak is seen in H2O when the (incoherent) neutron scattering susceptibility is measured at Q=7 nm −1. The Q dependence of the peak position demonstrates that dipolar relaxation is linked to molecular diffusion and gives the length scale of collective dipolar relaxation. The researchers deduce that the 8.3-ps dynamics is due to the movement of atoms along relatively large distances (0.34 nm), which are comparable to or slightly larger than intermolecular distances.
Similarly to the study of dielectric relaxation [2], the authors’ neutron scattering experiment also reveals two smaller peaks in the THz domain, a high-frequency range hardly accessible by dielectric relaxation. Because their position doesn't change with Q, the peaks correspond to localized motions. The peak at the highest frequency (around 2 THz) is associated with bending of chains of three oxygen molecules connected by hydrogen bonds (O—O—O). The other one, at intermediate frequencies (around 0.15 THz), is a novel finding of this work. This peak is related to local processes—motions of hydrogen atoms that take place when intermolecular bonds break and reform on picosecond time scales. By separating the contributions of oxygen and hydrogen atoms to scattering data, the researchers show that, on 0.1 to 1 ps time scales, the hydrogen atom moves in a “cage” with a size of 0.05 nm. Such a size corresponds to an amplitude of vibration or delocalization of hydrogens much smaller than the intermolecular distance, which is 0.28 nm on average.
These results confirm the presence of two dynamic molecular processes in liquid water, which are related to the diffusion of molecules and local movements associated with the making and breaking of hydrogen bonds, respectively. As the authors pointed out, this picture is similar to that observed in polymers [3] and also theoretically proposed for liquid water [4], which involves two main types of relaxation (called 𝛼 and 𝛽) taking place on different spatial and temporal scales.
An important aspect of this work is the fact that its conclusions do not rely on heavy simulations or complex computer models of water but are solely drawn from the experimental data. The natural continuation of Arbe et al.’s work should be the study of other temperatures. Since 𝛼 and 𝛽 relaxation processes are expected to be affected very differently by temperature, the temperature dependence will help researchers separate the molecular dynamics related to each of the processes, which will be key to explaining the unique behavior of water.
This research is published in Physical Review Letters.
References
- A. Arbe, P. Malo de Molina, F. Alvarez, B. Frick, and J. Colmenero, “Dielectric Susceptibility of Liquid Water: Microscopic Insights from Coherent and Incoherent Neutron Scattering,” Phys. Rev. Lett. 117, 185501 (2016).
- N. Q. Vinh, M. S. Sherwin, S. J. Allen, D. K. George, A. J. Rahmani, and K. W. Plaxco, “High-Precision Gigahertz-to-Terahertz Spectroscopy of Aqueous Salt Solutions as a Probe of the Femtosecond-to-Picosecond Dynamics of Liquid Water,” J. Chem. Phys. 142, 164502 (2015).
- D. Richter, M. Monkenbusch, A. Arbe, J. Colmenero, and B. Farago, “Dynamic Structure Factors Due to Relaxation Processes in Glass-Forming Polymers,” Physica B 241, 1005 (1997).
- J. Teixeira, A. Luzar, and S. Longeville, “Dynamics of Hydrogen Bonds: How to Probe their Role in the Unusual Properties of Liquid Water,” J. Phys. Condens. Matter 18, S2353 (2006); J. Swenson and J. Teixeira, “The Glass Transition and Relaxation Behavior of Bulk Water and a Possible Relation to Confined Water,” J. Chem. Phys. 132, 014508 (2010).